Bioassays sind an jeder Phase der Arzneimittelentwicklung beteiligt, angefangen bei der Zielidentifizierung bis hin zur Entdeckung der Leitverbindung. Bioassays liefern wertvolle Informationen, die die therapeutische Wirksamkeit eines untersuchten Arzneimittels aufzeigen.
Die bei Bioassays generierten Daten spielen auch eine wichtige Rolle bei der Arzneimittelentwicklung und der Qualitätskontrolle fertiger biologischer Produkte. Richtig konzipierte Bioassays helfen bei der Beurteilung der biologischen Wirkung, Aktivität, des Signaltransduktionsprozesses und der Rezeptorbindungsfähigkeit eines Arzneimittels oder Biologikums an einem biologischen Ziel (Proteinen) im Vergleich zu einer Referenz oder einem Standard in einem geeigneten biologischen System.
Die an der Entdeckung und Entwicklung von Arzneimitteln beteiligten Pharma- und Biotechnologieunternehmen stehen ständig vor der Herausforderung, biologisch relevante Tests zur Analyse mehrerer potenzieller Mechanismen zu entwickeln.
Der Prozess umfasst die Verwendung von qualitätskritischen Reagenzien, die Verwendung spezifischer Zelllinien sowie gereinigter Testarzneimittel und Referenzarzneimittelprodukte, was manchmal zu einer Einschränkung führen kann. Die meisten dieser Aktivitäten benötigen ausreichend Zeit, was für Biopharma-Hersteller zu einem limitierenden Faktor werden kann.
Es lohnt sich, Aktivitäten auszulagern renommierte CRO-Dienstleister um Zeit bei den Entwicklungsbemühungen zu sparen und auch eine unvoreingenommene Meinung über die funktionellen Aktivitäten des Arzneimittels zu erhalten.
Die Veeda Group verfügt über qualifizierte und erfahrene Wissenschaftler, die Bioassays für Unternehmen entwerfen, entwickeln, durchführen und validieren und erstklassige Bioassay-Dienstleistungen anbieten (in vitro und in vivo), die aussagekräftige Daten generieren, um Pharma- und Biotechunternehmen auf ihrem Weg zur Arzneimittelentdeckung und -entwicklung zu unterstützen.
Zu den Erfahrungen der Veeda Group in der Entwicklung und Durchführung von Bioassays gehören:
- Plaque-Reduktions-Neutralisationstest (PRNT-Assay)
- In-vitro- Hautsensibilisierungstest zur Aktivierung menschlicher Zelllinien (h-CLAT-Assay)
- Nab-Test
- Assay-Entwicklung (Pharmakodynamik, Pharmakokinetik, Immunogenität und Biomarker-Bewertung)
- In Vivo Bioassays für Arzneimittelmoleküle wie Luteinisierendes Hormon, Epoetin, HCG, rekombinantes FSH, β-HCG und Insulin.
- ADCC-Assay für Biosimilars und verschiedene andere Assays wie Ex-vivo Assay, zellbasierter Assay, Rezeptorbindungsassay, Zytokinfreisetzungsassay und ADA-Assay.
Die Veeda Group bietet mit ihren zahlreichen Technologieplattformen integrierte Entdeckungs-, Entwicklungs- und Regulierungsdienste:
- Explorative toxikologische Studien
- Studien zur regulatorischen Toxikologie
- In vitro Biotests
- Ex-vivo Biotests
Die Gruppe verfügt außerdem über die Erfahrung im Umgang mit einer Vielzahl von Biotherapeutika wie therapeutischen monoklonalen Antikörpern, Insulin und Insulinanaloga, Zytokinen, Heparinen mit niedrigem Molekulargewicht, Biosimilars, Hormone und Biomarker.
Die Veeda-Gruppe hat gezeigt, dass sie in der Lage ist, rekombinante Proteine wie nicht-glykosylierte Proteine und Glykoproteine zu entwickeln, die aus bakteriellen oder Säugetier-Wirts-Expressionssystemen stammen.
Bioassays in der präklinischen Arzneimittelentwicklung
Biologische Tests oder Bioassays sind wesentliche Werkzeuge in präklinische Arzneimittelentwicklung. Präklinische Bioassays können sein in vivo, ex vivo und in vitro.
In vivo Bioassays ermöglichen eine realistischere und prädiktivere Messung der funktionellen Auswirkungen von Tests mit Referenzarzneimitteln oder Standardmaterial definierter Wirksamkeit, zusammen mit der Anwendung statistischer Tools, studienspezifischer Labortechniken und der Einhaltung des gut konzipierten Studienprotokolls.
Diese Tests erfassen die Komplexität der Zielbindung, des Metabolismus und der Pharmakokinetik neuartiger Arzneimittel besser als in vitro Bioassays.
Die am häufigsten verwendeten Versuchssäugetiere in-vivo Wirksamkeitstests sind Mäuse und Ratten. Gelegentlich können je nach Empfindlichkeit und Eignung der Tests auch andere Arten verwendet werden.
Entwicklung und Validierung von Bioassays
Bioassays werden als Screening-Methode verwendet, um die Signale zu identifizieren, die auf die gewünschte biologische Aktivität einer Reihe von Verbindungen hinweisen. Im Allgemeinen können durch einen Bioassay zwei verschiedene Arten von Signalen erzeugt werden: eine lineare Dosis-Wirkungs-Beziehung und eine sigmoidale (S-förmige) Dosis-Wirkungs-Beziehung.
Da eine Lösung nicht für alle Bioassays geeignet ist, ist es sinnvoll, die Daten auszuwerten und zu analysieren, um einen präzisen Ansatz für die Durchführung jedes Bioassays zu entwickeln.
Die Lebenszyklusphasen eines Bioassays sind unterteilt in:
Stufe 1: Methodendesign, -entwicklung und -optimierung
Stufe 2: Qualifizierung der Verfahrensleistung
Stufe 3: Überprüfung der Verfahrensleistung (für den Zweck geeignet)
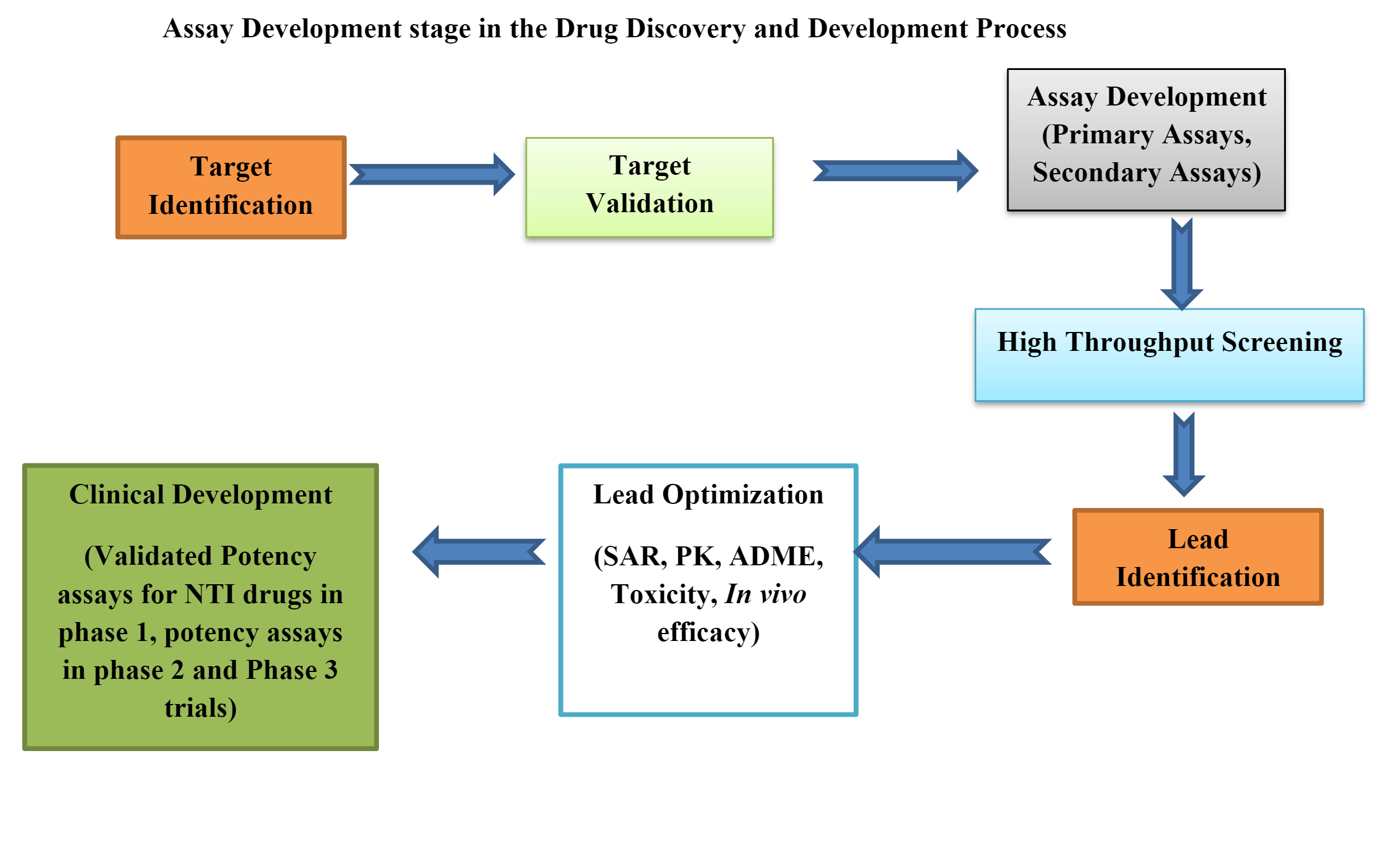
Die Entwicklung eines Bioassays, der die gesetzlichen Anforderungen erfüllt und die Registrierung eines Arzneimittels ermöglicht, ist ein sehr komplexer Prozess.
Die Entwicklung eines Bioassays umfasst viele Strategien und taktische Designs wie die Auswahl des richtigen in vivo Plattform, richtiges Methoden- oder Plattendesign, Datenanalyse, System-/Proben-Nachhaltigkeitsstrategie, Methodenimplementierung, Methodenleistung und Überwachung.
Für die Entwicklung und Validierung von Bioassays müssen mehrere Schritte befolgt werden, wie z. B. Auswahl von Dosis-Wirkungs- und Kurvenanpassung, Entwicklung einer Referenz, Berechnung der Wirksamkeit, Bioassay-Charakterisierung, Entwurf eines Bioassay-Rechners, Standardisierung und Automatisierung von Bioassays und schließlich , Auswertung.
Sowohl die Methodenentwicklung als auch die Validierung von Bioassays umfassen drei grundlegende Bereiche:
- Validierung vor der Studie (Identifizierungs- und Designphase).
- Validierung im Studium (Entwicklungs- und Produktionsphase).
- Kreuzvalidierung oder Methodentransfervalidierung
Während der Methodenentwicklung werden Testbedingungen und -verfahren ausgewählt, die die Auswirkungen potenzieller Ungültigkeitsquellen minimieren. Kommen wir zur statistischen Validierung für eine in vivo Es umfasst vier Hauptkomponenten:
- Angemessenes Studiendesign und Datenanalysemethode
- Richtige Randomisierung der Tiere
- Angemessene statistische Aussagekraft und Stichprobengröße
- Ausreichende Reproduzierbarkeit über alle Testläufe hinweg.
Parallelgruppendesign, randomisierte Blockdesign, Design mit wiederholten Messungen und Crossover-Design sind die grundlegenden Arten experimenteller Designs, die in verwendet werden in vivo Assay.
Im Folgenden sind die Schlüsselfaktoren aufgeführt, die beim Entwerfen eines berücksichtigt werden sollten in vivo Test:
- Alle (pharmakologisch) bedeutsamen biologischen Wirkungen sollten statistisch signifikant sein.
- Liegen keine biologisch relevanten Tests vor, kann eine Reihe plausibler Auswirkungen in Betracht gezogen werden.
- Die wichtigsten Endpunkte sollten vor Beginn des Tests genau definiert sein.
- Die Tiere sollten in geeigneter Weise nach dem Zufallsprinzip den Behandlungsgruppen zugeordnet werden.
- Die Dosierung sollte entsprechend gewählt werden. Die Auswahl der Dosis und der Kurvenanpassung gehört zu den kritischsten Aspekten der Bioassay-Entwicklung. Die Dosis wird abhängig von der Art des Modells bestimmt, das im Signal zur Anpassung an die Daten verwendet wird. Bei sigmoidalen Designs passt ein Vier- oder Fünf-Parameter-Logistikmodell (4PL oder 5PL) die Daten an, während bei linearem Design ein Parallellinienanalysemodell (PLA) an die Daten angepasst wird.
Für ein 4PL-Modell werden neun Dosen empfohlen:
- Drei Dosen in der unteren Asymptote
- Drei Dosen in der oberen Asymptote
- Drei Dosen im linearen Bereich
Im Gegensatz dazu werden für ein PLA-Modell mindestens vier Dosen empfohlen. Zur Darstellung der Dosiskurve sind mindestens drei aufeinanderfolgende Dosen erforderlich.
- Die Auswahl der Kontrollgruppen und Zeitpunkte für die Probenentnahme sollte optimal sein.
- Die Designstrategien sollten die Variabilität minimieren und die Informationen maximieren.
Um das Design, die Entwicklungen und die statistische Validierung zu verstehen in vivo Weitere Informationen zum Bioassay erhalten Sie unter https://www.veedacr.com. Sie können die vom NIH genannten Richtlinien auch lesen, indem Sie den Link besuchen:
https://www.ncbi.nlm.nih.gov/books/NBK92013/pdf/Bookshelf_NBK92013.pdf
Bibliographie
- A. Little, „Essentials in Bioassay Development“, BioPharm International 32 (11) 2019
- Padmalayam, Ph.D., Assay-Entwicklung in der Arzneimittelforschung
- Zwierzyna M, Overington JP (2017) Klassifizierung und Analyse einer großen Sammlung von In-vivo-Bioassay-Beschreibungen. PLoS Comput Biol13(7): e1005641. https://doi.org/10.1371/journal.pcbi.1005641
- White JR, Abodeely M, Ahmed S, Debauve G, Johnson E, Meyer DM, Mozier NM, Naumer M, Pepe A, Qahwash I, Rocnik E, Smith JG, Stokes ES, Talbot JJ, Wong PY. Best Practices in der Bioassay-Entwicklung zur Unterstützung der Registrierung von Biopharmazeutika. Biotechniken. 2019 Sep;67(3):126-137. doi: 10.2144/btn-2019-0031. Epub 2019. August 5. PMID: 31379198.
- F Chana und Hursh D, Bioassays während des Produktlebenszyklus: Perspektiven von CDER- und CBER-Reviews.
- Haas J, Manro J, Shannon H, et al. Richtlinien für In-vivo-Assays. 2012. Mai 1 [Aktualisiert am 2012. Okt. 1]. In: Markossian S., Grossman A., Brimacombe K. et al., Herausgeber. Assay Guidance Manual [Internet]. Bethesda (MD): Eli Lilly & Company und das National Center for Advancing Translational Sciences; 2004-. Bücherregal-URL: https://www.ncbi.nlm.nih.gov/books/