Testy biologiczne są stosowane na każdym etapie odkrywania leku, począwszy od identyfikacji celu aż do odkrycia związku wiodącego. Testy biologiczne dostarczają cennych informacji, które ukazują terapeutyczną moc badanego leku.
Dane generowane podczas testów biologicznych odgrywają również kluczową rolę w opracowywaniu leków i kontroli jakości gotowych produktów biologicznych. Prawidłowo zaprojektowane testy biologiczne pomagają w ocenie efektu biologicznego, aktywności, procesu przekazywania sygnału i zdolności wiązania receptora produktu leczniczego lub leku biologicznego z celem biologicznym (białkami) w porównaniu z punktem odniesienia lub standardem w odpowiednim systemie biologicznym.
Firmy farmaceutyczne i biotechnologiczne zaangażowane w odkrywanie i rozwój leków stoją przed ciągłym wyzwaniem związanym z opracowywaniem biologicznie istotnych testów do analizy wielu potencjalnych mechanizmów.
Proces ten obejmuje użycie odczynników o krytycznym znaczeniu dla jakości, użycie określonych linii komórkowych oraz oczyszczonych leków testowych i leków referencyjnych, co czasami może stanowić ograniczenie. Większość tych działań wymaga wystarczającej ilości czasu, co może stać się czynnikiem ograniczającym dla producentów biofarmaceutyków.
Komu warto zlecać działania renomowanych dostawców usług CRO aby zaoszczędzić czas w wysiłkach rozwojowych, a także mieć bezstronną opinię na temat aktywności funkcjonalnej produktu leczniczego.
Grupa Veeda zatrudnia wykwalifikowanych i doświadczonych naukowców do projektowania, opracowywania, wykonywania i walidacji testów biologicznych dla firm oraz zapewnia najwyższej jakości usługi w zakresie testów biologicznych (in vitro i in vivo), które generują istotne dane pomagające firmom farmaceutycznym i biotechnologicznym w odkrywaniu i opracowywaniu leków.
Doświadczenie Grupy Veeda w opracowywaniu i wykonywaniu testów biologicznych obejmuje:
- Test neutralizacji redukcji płytki nazębnej (test PRNT)
- In Vitro Działanie uczulające na skórę Test aktywacji ludzkiej linii komórkowej (test h-CLAT)
- Próba Naba
- Rozwój testów (farmakodynamika, farmakokinetyka, immunogenność i ocena biomarkerów)
- W Vivo Testy biologiczne na cząsteczki leków, takie jak hormon luteinizujący, epoetyna, HCG, rekombinowany FSH, β-HCG i insulina.
- Test ADCC dla leków biopodobnych i różne inne testy, takie jak Z życia test komórkowy, test wiązania receptora, test uwalniania cytokin i test ADA.
Grupa Veeda świadczy zintegrowane usługi w zakresie odkrywania, rozwoju i regulacji w ramach wielu platform technologicznych:
- Eksploracyjne badania toksykologiczne
- Regulacyjne badania toksykologiczne
- In vitro Testy biologiczne
- Ex vivo Testy biologiczne
Grupa ma również doświadczenie w leczeniu różnorodną gamą leków bioterapeutycznych, takich jak terapeutyczne przeciwciała monoklonalne, insulina i analogi insuliny, cytokiny, heparyny drobnocząsteczkowe, Biosimilary, Hormony i biomarkery.
Grupa Veeda wykazała możliwości opracowania rekombinowanych białek, takich jak białka nieglikozylowane i glikoproteiny pochodzące z systemów ekspresyjnych gospodarza bakteryjnego lub ssaczego.
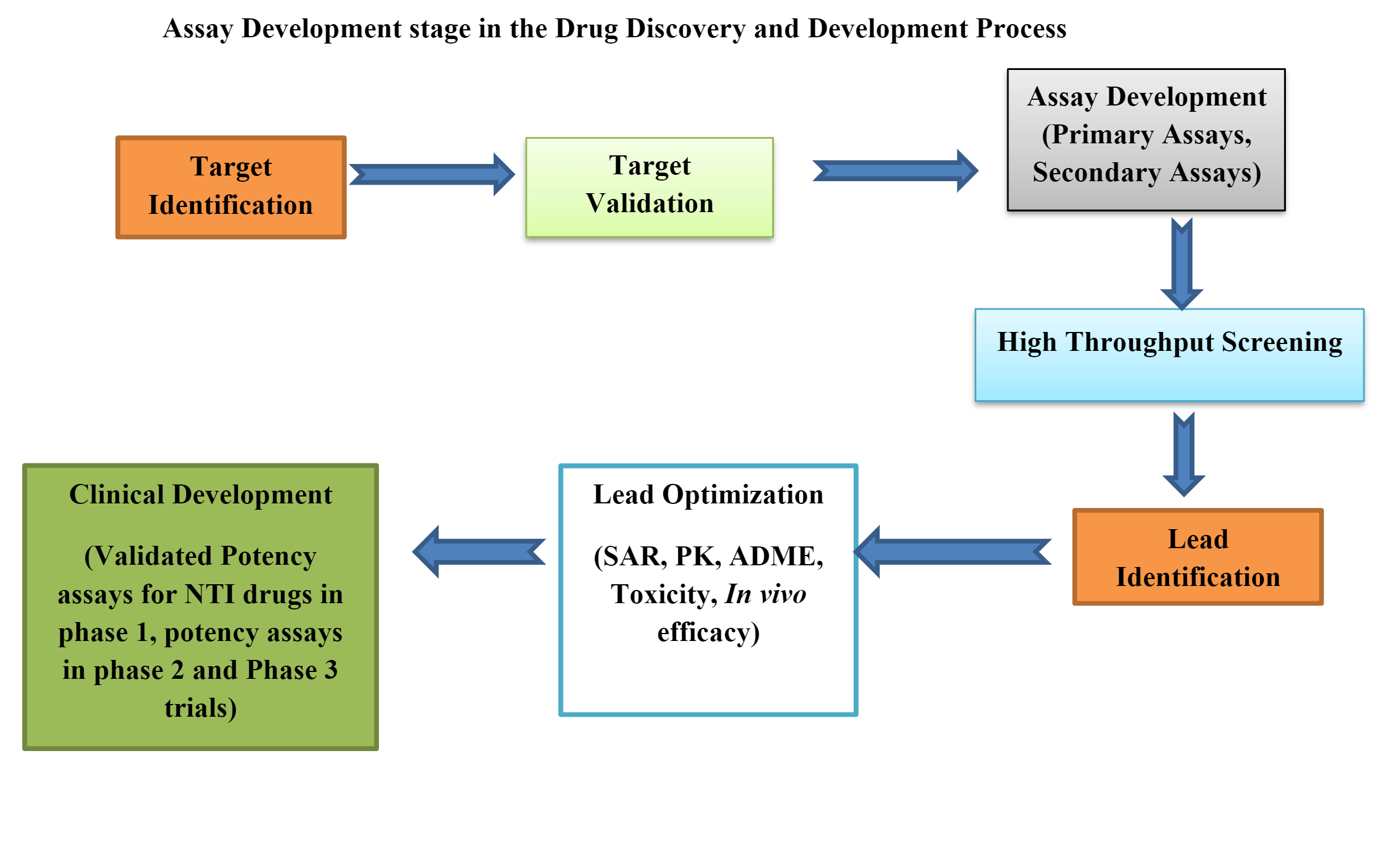
Testy biologiczne w przedklinicznym opracowywaniu leków
Testy biologiczne lub testy biologiczne są niezbędnymi narzędziami w przedkliniczny rozwój leków. Mogą być przedkliniczne testy biologiczne in vivo, ex vivo, in vitro.
In vivo testy biologiczne zapewniają bardziej realistyczny i predykcyjny pomiar efektów funkcjonalnych testów z referencyjnymi produktami leczniczymi lub standardowym materiałem o określonej sile, wraz z zastosowaniem narzędzi statystycznych, technik laboratoryjnych specyficznych dla badania i przestrzegania dobrze zaprojektowanego protokołu badania.
Testy te lepiej oddają złożoność działania docelowego, metabolizmu i farmakokinetyki nowych leków in vitro testy biologiczne.
Najczęściej wykorzystywane ssaki doświadczalne w m.inna żywo Testy skuteczności przeprowadzono na myszach i szczurach. Czasami można zastosować inne gatunki, w zależności od czułości i przydatności testów.
Opracowywanie i walidacja testów biologicznych
Testy biologiczne stosuje się jako metodę przesiewową w celu identyfikacji sygnałów wskazujących pożądaną aktywność biologiczną zestawu związków. Ogólnie rzecz biorąc, za pomocą testu biologicznego można wygenerować dwa różne typy sygnałów: liniową zależność dawka-odpowiedź i sigmoidalną (w kształcie litery S) odpowiedź na dawkę.
Ponieważ jedno rozwiązanie nie pasuje do wszystkich testów biologicznych, dobrze jest ocenić i przeanalizować dane, aby opracować precyzyjne podejście do przeprowadzenia każdego testu biologicznego.
Etapy cyklu życia testu biologicznego dzielą się na:
Etap 1: Projektowanie, rozwój i optymalizacja metody
Etap 2: Kwalifikacja wykonania procedury
Etap 3: Weryfikacja działania procedury (odpowiedni do celu)
Opracowanie testu biologicznego spełniającego wymogi regulacyjne i umożliwiającego rejestrację produktu leczniczego jest bardzo złożonym procesem.
Opracowanie testu biologicznego obejmuje wiele strategii i projektów taktycznych, takich jak wybór prawidłowego in vivo platforma, właściwy projekt metody lub płytki, analiza danych, strategia zrównoważonego rozwoju systemu/próbki, wdrożenie metody, działanie metody i monitorowanie.
Opracowanie i walidacja testów biologicznych obejmuje kilka etapów, takich jak wybór odpowiedzi na dawkę i dopasowanie krzywej, opracowanie punktu odniesienia, obliczenie siły działania, charakterystyka testu biologicznego, zaprojektowanie kalkulatora testu biologicznego, standaryzacja i automatyzacja testu biologicznego i wreszcie , ocena.
Zarówno rozwój metod, jak i walidacja testów biologicznych obejmują trzy podstawowe obszary:
- Walidacja przed badaniem (faza identyfikacji i projektowania).
- Walidacja w trakcie badania (faza rozwoju i produkcji).
- Walidacja krzyżowa lub walidacja transferu metod
Podczas opracowywania metody wybierane są warunki i procedury testu, które minimalizują wpływ potencjalnych źródeł nieważności. Przechodząc do walidacji statystycznej dla in vivo test obejmuje cztery główne elementy:
- Odpowiedni projekt badania i metoda analizy danych
- Właściwa randomizacja zwierząt
- Odpowiednia moc statystyczna i wielkość próby
- Odpowiednia odtwarzalność w seriach testów.
Projekt grup równoległych, projekt bloków losowych, projekt powtarzanych pomiarów i projekt krzyżowania to podstawowe typy projektów eksperymentów stosowanych w in vivo analiza.
Poniżej przedstawiono najważniejsze czynniki, o których należy pamiętać podczas projektowania in vivo analiza:
- Wszystkie znaczące skutki biologiczne (farmakologiczne) powinny być istotne statystycznie.
- Jeśli nie istnieją biologicznie istotne testy, można rozważyć szereg prawdopodobnych skutków.
- Kluczowe punkty końcowe powinny być dobrze zdefiniowane przed rozpoczęciem testu.
- Zwierzęta należy przydzielać losowo, w odpowiedni sposób, do grup poddawanych leczeniu.
- Poziomy dawek należy dobrać odpowiednio. Wybór dawki i dopasowania krzywej jest jednym z najważniejszych aspektów rozwoju testów biologicznych. Dawkę określa się w zależności od rodzaju modelu zastosowanego w sygnale w celu dopasowania go do danych. W przypadku projektów sigmoidalnych do danych pasuje cztero- lub pięcioparametrowy model logistyczny (4PL lub 5PL), natomiast w przypadku projektów liniowych do danych pasuje model analizy linii równoległych (PLA).
Dla modelu 4PL zaleca się dziewięć dawek:
- Trzy dawki w dolnej asymptocie
- Trzy dawki w górnej asymptocie
- Trzy dawki w zakresie liniowym
Natomiast w przypadku modelu PLA zalecane są minimum cztery dawki. Do wykreślenia krzywej dawki wymagane są co najmniej trzy kolejne dawki.
- Wybór grup kontrolnych i punktów czasowych pobrania próbek powinien być optymalny.
- Strategie projektowania powinny minimalizować zmienność i maksymalizować ilość informacji.
Aby zrozumieć projekt, rozwój i walidację statystyczną in vivo bardziej szczegółowo, skontaktuj się z nami pod adresem https://www.veedacr.com. Z wytycznymi wspomnianymi przez NIH można także zapoznać się klikając na link:
https://www.ncbi.nlm.nih.gov/books/NBK92013/pdf/Bookshelf_NBK92013.pdf
Referencje
- A. Little, „Essentials in Bioassay Development”, BioPharm International 32 (11) 2019
- Doktor Padmalayam, rozwój testów w odkrywaniu leków
- Zwierzyna M, Overington JP (2017) Klasyfikacja i analiza dużego zbioru opisów testów biologicznych in vivo. PLoS Comput Biol13(7): e1005641. https://doi.org/10.1371/journal.pcbi.1005641
- White JR, Abodeely M, Ahmed S, Debauve G, Johnson E, Meyer DM, Mozier NM, Naumer M, Pepe A, Qahwash I, Rocnik E, Smith JG, Stokes ES, Talbot JJ, Wong PY. Najlepsze praktyki w opracowywaniu testów biologicznych wspierających rejestrację produktów biofarmaceutycznych. Biotechniki. wrzesień 2019;67(3):126-137. doi: 10.2144/btn-2019-0031. EPUB 2019, 5 sierpnia. PMID: 31379198.
- F Chana i Hursh D, Testy biologiczne w cyklu życia produktu: Perspektywy przeglądów CDER i CBER.
- Haas J., Manro J., Shannon H. i in. Wytyczne dotyczące testów in vivo. 2012 maja 1 r. [Aktualizacja: 2012 października 1 r.]. W: Markossian S, Grossman A, Brimacombe K i in., wyd. Podręcznik wytycznych dotyczących testów [Internet]. Bethesda (MD): Eli Lilly & Company i Krajowe Centrum Rozwoju Nauk Translacyjnych; 2004-. Adres URL półki na książki: https://www.ncbi.nlm.nih.gov/books/