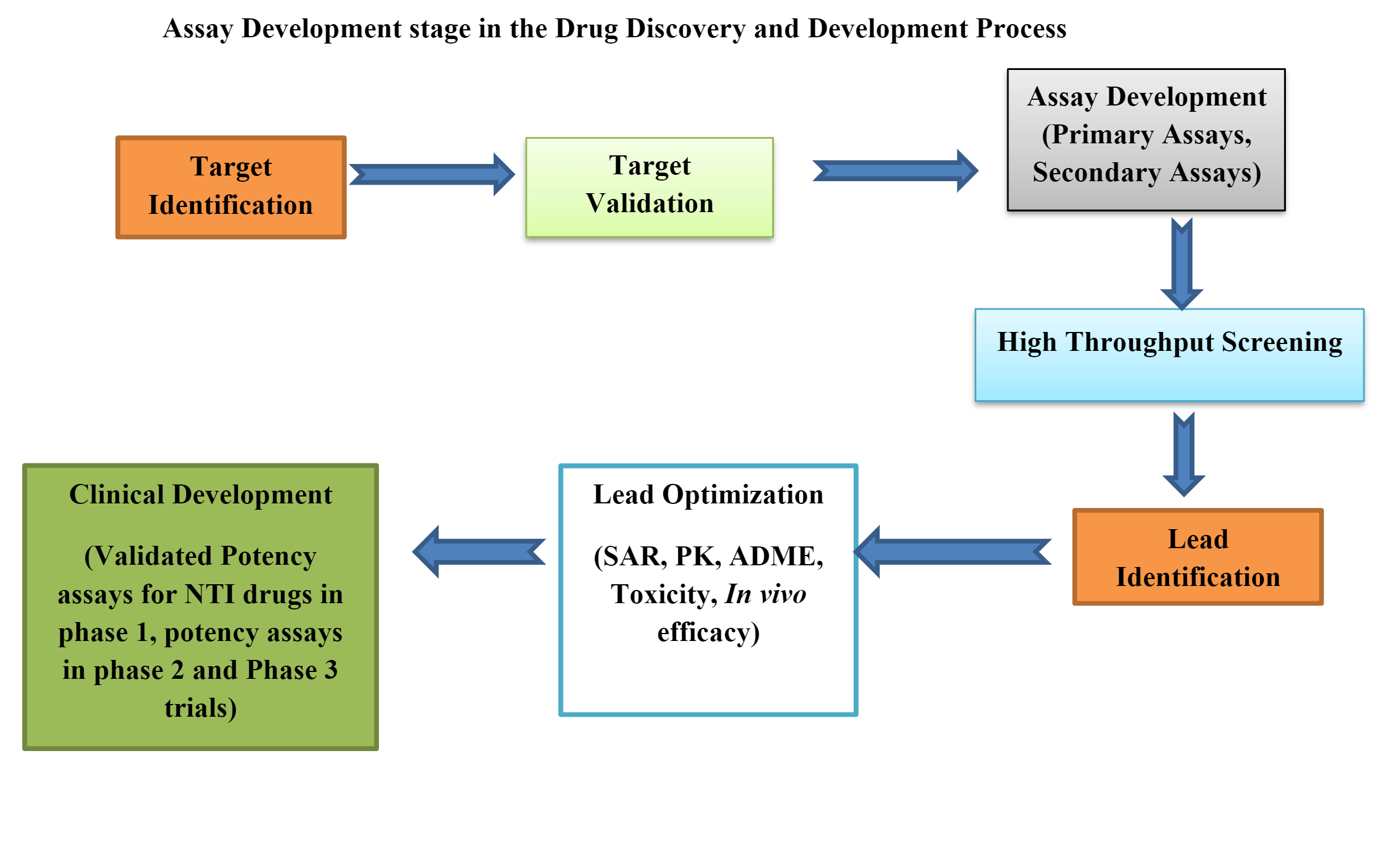
Les essais biologiques sont impliqués à chaque étape de la découverte d'un médicament, depuis l'identification de la cible jusqu'à la découverte du composé principal. Les essais biologiques fournissent des informations précieuses qui démontrent la puissance thérapeutique d'un médicament faisant l'objet d'une enquête.
Les données générées lors des essais biologiques jouent également un rôle essentiel dans le développement de médicaments et le contrôle qualité des produits biologiques finis. Des essais biologiques correctement conçus aident à évaluer l'effet biologique, l'activité, le processus de transduction du signal et la capacité de liaison au récepteur d'un produit médicamenteux ou d'un produit biologique sur une cible biologique (protéines) par rapport à une référence ou un standard sur un système biologique approprié.
Les sociétés pharmaceutiques et biotechnologiques impliquées dans la découverte et le développement de médicaments sont continuellement confrontées au défi de développer des tests biologiquement pertinents pour l'analyse de multiples mécanismes potentiels.
Le processus implique l’utilisation de réactifs de qualité critique, l’utilisation de lignées cellulaires spécifiques et de médicaments d’essai purifiés et de produits médicamenteux de référence, ce qui peut parfois devenir une contrainte. La plupart de ces activités nécessitent suffisamment de temps, ce qui peut devenir un facteur limitant pour les fabricants biopharmaceutiques.
Cela vaut la peine d'externaliser les activités Fournisseurs de services CRO réputés pour gagner du temps dans les efforts de développement et également pour avoir une opinion impartiale sur les activités fonctionnelles du produit médicamenteux.
Le groupe Veeda dispose de scientifiques qualifiés et expérimentés pour concevoir, développer, exécuter et valider les essais biologiques pour les entreprises et fournit des services d'essais biologiques de premier ordre (in vitro ainsi que in vivo) qui génèrent des données significatives pour soutenir les entreprises pharmaceutiques et biotechnologiques dans leur parcours de découverte et de développement de médicaments.
L'expérience du groupe Veeda dans le développement et l'exécution d'essais biologiques comprend :
- Test de neutralisation de la réduction des plaques (test PRNT)
- In vitro Test d'activation de lignées cellulaires humaines de sensibilisation cutanée (test h-CLAT)
- Test Nab
- Développement de tests (pharmacodynamique, pharmacocinétique, immunogénicité et évaluation des biomarqueurs)
- In vivo Essais biologiques pour des molécules médicamenteuses telles que l'hormone lutéinisante, l'époétine, l'HCG, la FSH recombinante, la β-HCG et l'insuline.
- Test ADCC pour les biosimilaires et différents autres tests comme Ex vivo test, test cellulaire, test de liaison aux récepteurs, test de libération de cytokines et test ADA.
Veeda Group fournit des services intégrés de découverte, de développement et de réglementation avec ses multiples plateformes technologiques :
- Etudes exploratoires de toxicologie
- Études de toxicologie réglementaire
- In vitro Essais biologiques
- Ex vivo Essais biologiques
Le groupe possède également l'expérience nécessaire pour gérer une gamme diversifiée de produits biothérapeutiques tels que les anticorps monoclonaux thérapeutiques, l'insuline et ses analogues, les cytokines, les héparines de faible poids moléculaire, Biosimilaires, Hormones et biomarqueurs.
Le groupe Veeda a démontré sa capacité à développer des protéines recombinantes telles que des protéines non glycosylées et des glycoprotéines dérivées de systèmes d'expression d'hôtes bactériens ou mammifères.
Essais biologiques dans le développement de médicaments précliniques
Les analyses biologiques ou essais biologiques sont des outils essentiels dans développement préclinique de médicaments. Les essais biologiques précliniques peuvent être in vivo, ex vivoet in vitro.
in vivo les essais biologiques fournissent une mesure plus réaliste et prédictive des effets fonctionnels des tests avec des produits médicamenteux de référence ou du matériel standard d'activité définie, ainsi que l'application d'outils statistiques, de techniques de laboratoire spécifiques à l'étude et le respect du protocole d'étude bien conçu.
Ces tests capturent mieux la complexité de l'engagement des cibles, du métabolisme et de la pharmacocinétique des nouveaux médicaments que in vitro bioessais.
Les mammifères expérimentaux les plus couramment utilisés en iin vivo les tests d'efficacité sont des souris et des rats. Occasionnellement, d’autres espèces peuvent être utilisées en fonction de la sensibilité et de l’adéquation des tests.
Développement et validation d'essais biologiques
Les essais biologiques sont utilisés comme méthode de criblage pour identifier les signaux qui indiquent l'activité biologique souhaitée à partir d'un ensemble de composés. En général, deux types différents de signaux peuvent être générés par un essai biologique, une dose-réponse linéaire et une dose-réponse sigmoïde (en forme de S).
Puisqu’une solution unique ne convient pas à tous les essais biologiques, il est bon d’évaluer et d’analyser les données afin de développer une approche précise pour réaliser chaque essai biologique.
Les étapes du cycle de vie d’un essai biologique sont divisées en :
Étape 1 : Conception, développement et optimisation de la méthode
Étape 2 : Qualification de la performance de la procédure
Étape 3 : Vérification de la performance de la procédure (adapté à l'objectif)
Développer un essai biologique qui répond aux exigences réglementaires et permet d’enregistrer un produit médicamenteux est un processus très complexe.
L'élaboration d'un essai biologique implique de nombreuses stratégies et conceptions tactiques, comme la sélection du bon in vivo plate-forme, conception appropriée de la méthode ou de la plaque, analyse des données, stratégie de durabilité du système/échantillon, mise en œuvre de la méthode, performances de la méthode et surveillance.
Il y a plusieurs étapes à suivre pour le développement et la validation des essais biologiques, telles que la sélection dose-réponse et d'ajustement de courbe, le développement de références, le calcul de l'activité, la caractérisation des essais biologiques, la conception du calculateur d'essais biologiques, la normalisation et l'automatisation des essais biologiques, et enfin , évaluation.
Le développement de méthodes et la validation des essais biologiques comprennent trois domaines fondamentaux :
- Validation pré-étude (phase d’identification et de conception)
- Validation en cours d'étude (phase de développement et de production)
- Validation croisée ou validation de transfert de méthode
Lors du développement de la méthode, les conditions et procédures d'analyse sont sélectionnées pour minimiser l'impact des sources potentielles d'invalidité. En venant à la validation statistique d'un in vivo test, il implique quatre composants principaux :
- Conception d’étude et méthode d’analyse des données adéquates
- Randomisation appropriée des animaux
- Puissance statistique et taille d'échantillon appropriées
- Reproductibilité adéquate entre les analyses.
Le plan en groupes parallèles, le plan en blocs randomisés, le plan à mesures répétées et le plan croisé sont les types de base de plans expérimentaux utilisés dans in vivo essai.
Voici les facteurs clés à garder à l’esprit lors de la conception d’un in vivo essai:
- Tous les effets biologiques significatifs (pharmacologiquement) doivent être statistiquement significatifs.
- En l’absence de tests biologiquement pertinents, une gamme d’effets plausibles peut être envisagée.
- Les paramètres clés doivent être bien définis avant le début du test.
- Les animaux doivent être répartis au hasard et de manière appropriée dans les groupes de traitement.
- Les niveaux de dose doivent être sélectionnés de manière appropriée. La sélection de la dose et de l’ajustement de la courbe fait partie des aspects les plus critiques du développement d’essais biologiques. La dose est déterminée en fonction du type de modèle utilisé dans le signal pour ajuster les données. Pour les conceptions sigmoïdales, un modèle logistique à quatre ou cinq paramètres (4PL ou 5PL) ajuste les données, tandis que, pour la conception linéaire, un modèle d'analyse de lignes parallèles (PLA) ajuste les données.
Pour un modèle 4PL, neuf doses sont recommandées :
- Trois doses dans l'asymptote inférieure
- Trois doses dans l'asymptote supérieure
- Trois doses dans la plage linéaire
En revanche, pour un modèle PLA, un minimum de quatre doses est recommandé. Un minimum de trois doses consécutives est requis pour tracer la courbe de dose.
- La sélection des groupes témoins et des moments de collecte des échantillons doit être optimale.
- Les stratégies de conception doivent minimiser la variabilité et maximiser les informations.
Comprendre la conception, les développements et la validation statistique de in vivo essai biologique plus en détail, contactez-nous à https://www.veedacr.com. On peut également lire les lignes directrices mentionnées par le NIH en visitant le lien :
https://www.ncbi.nlm.nih.gov/books/NBK92013/pdf/Bookshelf_NBK92013.pdf
Bibliographie
- A. Little, « Éléments essentiels du développement de bioessais », BioPharm International 32 (11) 2019
- Padmalayam, Ph.D., Développement de tests pour la découverte de médicaments
- Zwierzyna M, Overington JP (2017) Classification et analyse d'une vaste collection de descriptions d'essais biologiques in vivo. PLoS Comput Biol13(7) : e1005641. https://doi.org/10.1371/journal.pcbi.1005641
- White JR, Abodeely M, Ahmed S, Debauve G, Johnson E, Meyer DM, Mozier NM, Naumer M, Pepe A, Qahwash I, Rocnik E, Smith JG, Stokes ES, Talbot JJ, Wong PY. Meilleures pratiques en matière de développement d'essais biologiques pour soutenir l'enregistrement des produits biopharmaceutiques. Biotechniques. Septembre 2019;67(3):126-137. est ce que je : 10.2144/btn-2019-0031. Publication en ligne le 2019 août 5. PMID : 31379198.
- F Chana et Hursh D, Essais biologiques tout au long du cycle de vie du produit : perspectives des examens du CDER et du CBER.
- Haas J, Manro J, Shannon H et al. Directives pour les tests in vivo. 2012er mai 1 [Mise à jour le 2012er octobre 1]. Dans : Markossian S, Grossman A, Brimacombe K et al., éditeurs. Manuel d'orientation des analyses [Internet]. Bethesda (MD) : Eli Lilly & Company et le National Center for Advancing Translational Sciences ; 2004-. URL de la bibliothèque : https://www.ncbi.nlm.nih.gov/books/