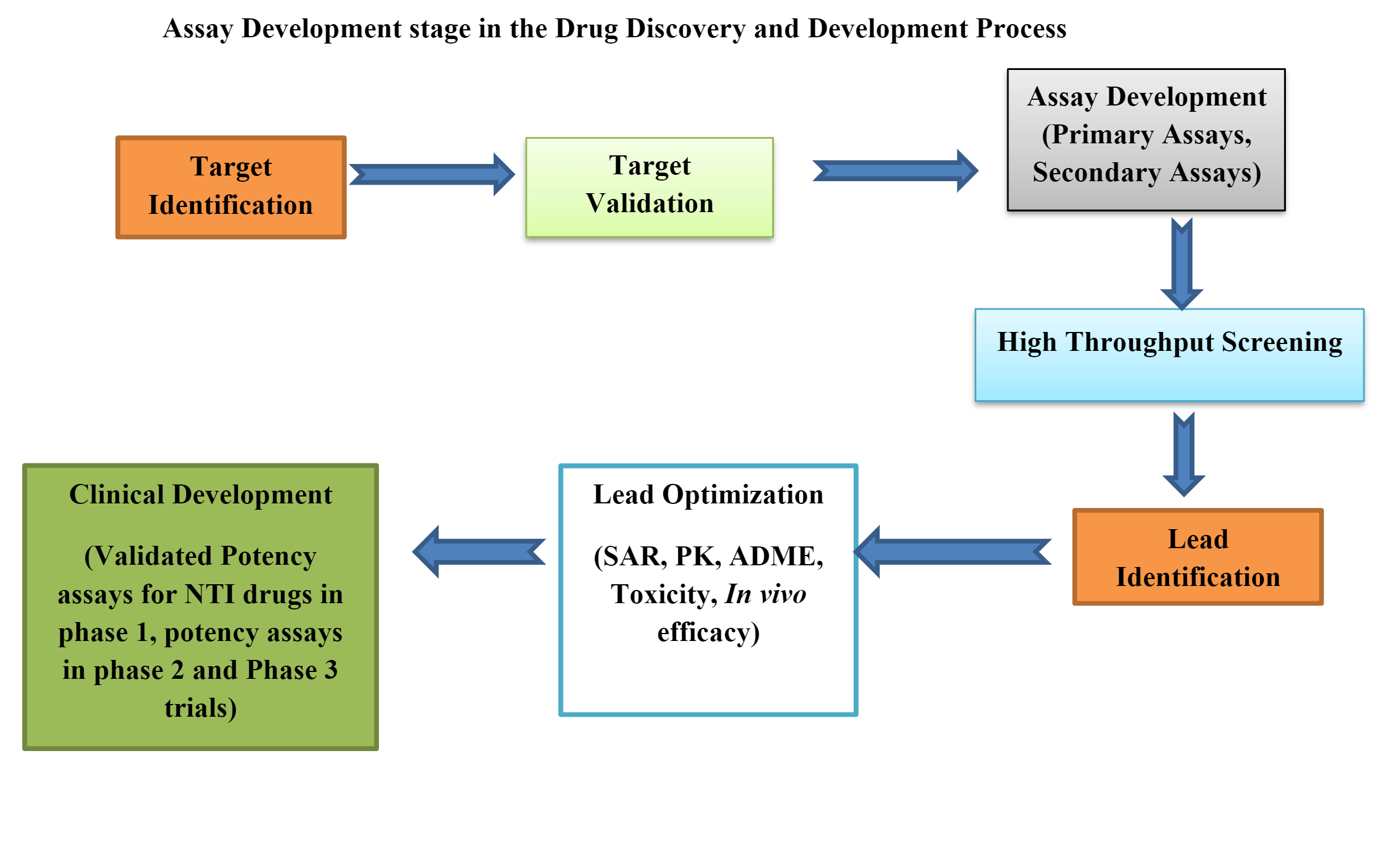
生物测定涉及药物发现的每个阶段,从靶标识别到发现先导化合物。 生物测定提供了有价值的信息,显示了正在研究的药物的治疗效力。
生物测定过程中产生的数据在药物开发和生物制品成品的质量控制中也发挥着至关重要的作用。 正确设计的生物测定有助于评估药物产品或生物制剂在生物靶标(蛋白质)上与适当生物系统的参考或标准相比的生物效应、活性、信号转导过程和受体结合能力。
参与药物发现和开发的制药和生物技术公司不断面临开发生物学相关检测方法来分析多种潜在机制的挑战。
该过程涉及质量关键试剂的使用、特定细胞系的使用以及纯化的测试药物和参考药物产品,这些有时可能成为限制。 这些活动大多数需要足够的时间,这可能成为生物制药制造商的限制因素。
值得将活动外包给 知名CRO服务商 节省开发时间,并对药品的功能活性有公正的看法。
Veeda 集团拥有合格且经验丰富的科学家为公司设计、开发、执行和验证生物测定,并提供一流的生物测定服务(细胞/组织 和 体内)生成有意义的数据,以支持制药和生物技术公司的药物发现和开发之旅。
Veeda 集团在生物测定开发和执行方面的经验包括:
- 斑块减少中和试验(PRNT 测定)
- 体外 皮肤致敏人类细胞系激活测试(h-CLAT 测定)
- 蛋白结合分析
- 检测开发(药效学、药代动力学、免疫原性和生物标志物评估)
- 体内 黄体生成素、依泊汀、HCG、重组 FSH、β-HCG 和胰岛素等药物分子的生物测定。
- 生物仿制药的 ADCC 测定和不同的其他测定,例如 离体 测定、基于细胞的测定、受体结合测定、细胞因子释放测定和 ADA 测定。
Veeda Group 通过其多个技术平台提供综合发现、开发和监管服务:
- 探索性毒理学研究
- 监管毒理学研究
- 细胞/组织 生物测定
- 离体 生物测定
该团队还拥有处理各种生物治疗药物的经验,例如治疗性单克隆抗体、胰岛素和胰岛素类似物、细胞因子、低分子量肝素、 生物仿制药、激素和生物标志物。
Veeda 集团已展示出开发重组蛋白的能力,例如源自细菌或哺乳动物宿主表达系统的非糖基化蛋白和糖蛋白。
临床前药物开发中的生物测定
生物测定或生物测定是重要的工具 临床前药物开发。 临床前生物测定可以 体内、离体及 细胞/组织.
体内 生物测定提供了对参考药物产品或确定效力的标准材料测试的功能效果的更现实和预测性的测量,以及统计工具的应用、研究特定的实验室技术以及对精心设计的研究方案的遵守。
这些检测方法比新药更好地捕捉了靶点参与、代谢和药代动力学的复杂性。 细胞/组织 生物测定。
我国最常用的实验哺乳动物体内 功效测定是小鼠和大鼠。 有时可能会使用其他物种,具体取决于测定的灵敏度和适用性。
生物测定的开发和验证
生物测定用作筛选方法,以从一组化合物中识别指示所需生物活性的信号。 一般来说,生物测定可以产生两种不同类型的信号:线性剂量响应和S形剂量响应。
由于一种解决方案并不适合所有生物测定,因此最好评估和分析数据以开发一种精确的方法来进行每种生物测定。
生物测定的生命周期阶段分为:
第一阶段:方法设计、开发和优化
第 2 阶段:程序性能鉴定
第 3 阶段:程序性能验证(适合目的)
开发满足监管要求并获得药品注册的生物测定方法是一个非常复杂的过程。
开发生物测定包括许多策略和战术设计,例如选择正确的 体内 平台、正确的方法或板设计、数据分析、系统/样品可持续性策略、方法实施、方法性能和监控。
生物测定的开发和验证需要遵循几个步骤,例如剂量反应和曲线拟合选择、参考文献的开发、效价计算、生物测定表征、生物测定计算器的设计、生物测定的标准化和自动化,最后, 评估。
生物测定的方法开发和验证都包括三个基本领域:
- 研究前(识别和设计阶段)验证
- 研究中(开发和生产阶段)验证
- 交叉验证或方法转移验证
在方法开发过程中,选择的检测条件和程序应尽量减少潜在无效来源的影响。 进行统计验证 体内 化验,它涉及四个主要组成部分:
- 充分的研究设计和数据分析方法
- 动物的适当随机化
- 适当的统计功效和样本量
- 整个检测运行具有足够的重现性。
平行组设计、随机区组设计、重复测量设计和交叉设计是实验设计的基本类型 体内 分析。
以下是设计时应牢记的关键因素 体内 分析:
- 所有有意义的生物效应(药理学)都应该具有统计学意义。
- 如果不存在生物学相关的测定,则可以考虑一系列可能的效应。
- 在检测开始之前应明确定义关键终点。
- 应以适当的方式将动物随机分配到治疗组。
- 应适当选择剂量水平。 剂量和曲线拟合选择是生物测定开发中最关键的方面之一。 根据信号中用于拟合数据的模型类型来确定剂量。 对于 S 形设计,四参数或五参数物流(4PL 或 5PL)模型适合数据,而对于线性设计,平行线分析 (PLA) 模型适合数据。
对于 4PL 模型,建议使用九剂:
- 下渐近线的三个剂量
- 上渐近线的三个剂量
- 线性范围内的三个剂量
相比之下,对于 PLA 模型,建议至少使用四剂。 绘制剂量曲线至少需要三个连续剂量。
- 对照组和收集样本的时间点的选择应该是最佳的。
- 设计策略应最小化可变性并最大化信息。
了解设计、开发和统计验证 体内 有关生物测定的更多详细信息,请联系我们 https://www.veedacr.com。 人们还可以通过访问以下链接阅读 NIH 提到的指南:
https://www.ncbi.nlm.nih.gov/books/NBK92013/pdf/Bookshelf_NBK92013.pdf
参考资料
- A. Little,“生物测定开发的要点”,BioPharm International 32 (11) 2019
- Padmalayam 博士,药物发现中的检测开发
- Zwierzyna M, Overington JP (2017) 对大量体内生物测定描述的分类和分析。 PLoS 计算机生物学 13(7):e1005641。 https://doi.org/10.1371/journal.pcbi.1005641
- White JR、Abodeely M、Ahmed S、Debauve G、Johnson E、Meyer DM、Mozier NM、Naumer M、Pepe A、Qahwash I、Rocnik E、Smith JG、Stokes ES、Talbot JJ、Wong PY。 支持生物制药注册的生物测定开发最佳实践。 生物技术。 2019年67月;3(126):137-10.2144。 doi:2019/btn-0031-2019。 Epub 5 年 31379198 月 XNUMX 日。PMID:XNUMX。
- F Chana 和 Hursh D,整个产品生命周期的生物测定:CDER 和 CBER 审查的观点。
- 哈斯 J、曼罗 J、香农 H 等人。 体内检测指南。 2012 年 1 月 2012 日 [1 年 2004 月 XNUMX 日更新]。 见:Markossian S、Grossman A、Brimacombe K 等人,编辑。 检测指导手册[互联网]。 贝塞斯达(医学博士):礼来公司和国家转化科学促进中心; XNUMX年-。 书架网址: https://www.ncbi.nlm.nih.gov/books/