Fortschritte in den medizinischen Wissenschaften haben der Menschheit in vielerlei Hinsicht Vorteile gebracht. Bei der Durchführung klinischer Studien wurden jedoch Vorfälle wissenschaftlichen, moralischen und ethischen Fehlverhaltens ans Licht gebracht, die die wissenschaftliche Gemeinschaft und die Öffentlichkeit erschüttert haben. Dies führte 1979 zur Gründung einer formellen Organisation durch die Vereinigten Staaten (USA), nämlich der „International Ethical Guidelines for Biomedical Research Involving Human Subjects“, um die Interessen der Versuchspersonen zu schützen und zu schützen. Daraufhin haben viele Länder ihre eigenen Richtlinien für gute klinische Praxis (Good Clinical Practices, GCP) entworfen. Da jedoch immer mehr klinische Studien an Standorten in mehreren Ländern durchgeführt werden, war es notwendig, einheitliche Richtlinien für die Durchführung klinischer Studien zu haben. Daraus entstanden 1996 die International Conference on Harmonization (ICH)-GCP-Richtlinien mit dem Ziel, einen einheitlichen Standard bereitzustellen, der die Akzeptanz klinischer Studiendaten durch die Regulierungsbehörden der jeweiligen Länder erleichtert. Im Laufe der Zeit haben viele Länder die ICH-GCP-Richtlinien angepasst, um ihre eigenen Richtlinien festzulegen. Auch Indien folgte diesem Beispiel und führte mit dem Indian Council of Medical Research (ICMR) die „Ethischen Richtlinien für die biomedizinische Forschung an Menschen“ ein, die kontinuierlich überarbeitet und ergänzt werden, um sicherzustellen, dass klinische Studien mit höchster Qualität durchgeführt werden und dem Wohlergehen der Menschen Vorrang eingeräumt wird beteiligte Themen.1
Indien – ein globales Reiseziel
Indien entwickelt sich aus mehreren Gründen zu einem bevorzugten Ziel für klinische Studien für viele internationale Unternehmen:
☉ Förderliches regulatorisches Umfeld: International harmonisierte und günstige Regulierungsprozesse wie die beschleunigte Zulassung neuer Prüfpräparate machen das klinische Forschungsumfeld in Indien für die Durchführung klinischer Studien zugänglicher. Markttrends zeigen eine durchschnittliche jährliche Wachstumsrate (CAGR) von etwa 12 % (987 Millionen US-Dollar) in der indischen Branche für klinische Studien, ausgehend von 500 Millionen US-Dollar im Jahr 2017.1,2,3,4,5
☉ Geschulte Arbeitskräfte: Verfügbarkeit qualifizierter medizinischer Fachkräfte, die auf verschiedene Therapiebereiche spezialisiert sind, sich gut mit der englischen Sprache auskennen und die Einhaltung der ICH-GCP-Richtlinien sicherstellen.1,2,3
☉ Technologieinfrastruktur: Weltklasse-Technologien im Datenmanagement und in der Informationstechnologie sowie damit verbundene Dienstleistungen.1,2,3
☉ Patientenpool: Große Bevölkerung, die behandlungsnaiv ist und eine vielfältige genetische und ethnische Zusammensetzung aufweist. Da Indien zunehmend urbanisiert wird und die Verbindung zwischen städtischen und ländlichen Gebieten immer besser wird, wird es einfacher, Patienten aus verschiedenen geografischen Gebieten zu rekrutieren. Darüber hinaus gibt es eine hohe Inzidenz und Prävalenz akuter und chronischer Erkrankungen aufgrund von Lebensstiländerungen, die zu Krankheiten wie Diabetes, Krebs usw. führen. Solche lebensstilbedingten Störungen eröffnen die Möglichkeit, weitere klinische Studien in diesen Krankheitsbereichen durchzuführen.1,2,3,6
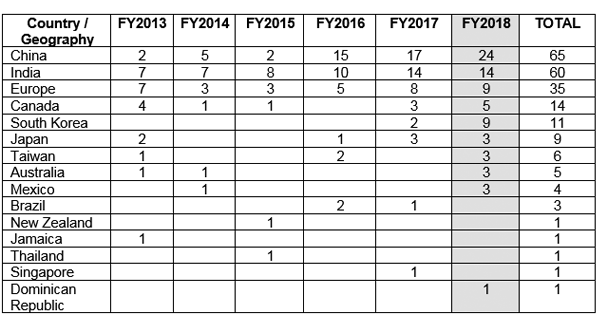
☉ Einfache Rekrutierung: In Ländern wie den USA gelingt es etwa 86 % der klinischen Studien nicht, die erforderliche Anzahl an Probanden zu rekrutieren, was zu einer Verzögerung von fast einem Jahr führt. Diese Verzögerung kostet das Sponsorunternehmen mehrere Millionen Dollar. Zu den Gründen für eine verspätete Rekrutierung zählen mangelnde Bereitschaft des Patienten zur Teilnahme, strenge Sicherheitsanforderungen und hohe Vergütungspakete. Indien bietet die Möglichkeit einer relativ einfachen Patientenrekrutierung aufgrund der verbesserten Studienkonformität und Transparenz, insbesondere durch die jüngste Veröffentlichung der New Drugs and Clinical Trial Rules 2019, die aktualisierte Regeln und Vorschriften für die schnelle Genehmigung klinischer Studien enthält. Unter den Ländern mit schnell wachsenden Volkswirtschaften wurde festgestellt, dass Indien eine Wachstumsrate bei den Rekrutierungsstandorten von etwa 22.6 % aufweist, wobei die höchste Wachstumsrate in China zu verzeichnen ist (≈36 %).1,2,7,8
☉ Wettbewerbsfähige Kosten – Die Kosteneffizienz ist ein ausschlaggebender Faktor dafür, dass viele Studien nach Indien verlagert werden. Schätzungen zufolge liegen die Kosten für die Entwicklung eines neuen Medikaments fast 50 % unter denen, die in den USA oder der Europäischen Union erforderlich wären. 1,2,3
Zukunft der klinischen Forschung in Indien
Die Central Drugs Standard Control Organization (CDSCO) arbeitet derzeit an spezifischen Richtlinien für Stammzellenforschung, Biosimilars und medizinische Geräte, um Patienten zu schützen und die klinische Forschung und Entwicklung im Land zu fördern. Nach einer Flaute in der indischen klinischen Industrie vor 2015 aufgrund ethischer und qualitativer Bedenken hat die offene Kommunikation zwischen Sponsorenvertretern und dem Regulierungsteam von CDSCO dazu beigetragen, Indien erneut als potenzielles globales Ziel für die Einschreibung einer vielfältigen Bevölkerung in klinische Studien zu überdenken Halten Sie sich strikt an die ICH-GCP-Richtlinien.6
Haftungsausschluss:
Die in diesem Artikel enthaltenen Informationen dienen ausschließlich der allgemeinen Orientierung zu Themen, die für den persönlichen Gebrauch des Lesers von Interesse sind, der die volle Verantwortung für deren Verwendung übernimmt. Dementsprechend werden die Informationen in diesem Artikel mit der Maßgabe bereitgestellt, dass der/die Autor(en) und der/die Herausgeber hierin nicht mit der Erbringung professioneller Ratschläge oder Dienstleistungen befasst sind. Daher sollte es nicht als Ersatz für die Beratung durch einen kompetenten Berater dienen. Bevor der Leser eine Entscheidung trifft oder Maßnahmen ergreift, sollte er immer einen professionellen Berater zu Rate ziehen, der sich auf den betreffenden Artikelbeitrag bezieht.
Obwohl alle Anstrengungen unternommen wurden, um sicherzustellen, dass die in diesem Artikel enthaltenen Informationen aus zuverlässigen Quellen stammen, ist Veeda Clinical Research nicht für etwaige Fehler oder Auslassungen oder für die Ergebnisse verantwortlich, die sich aus der Verwendung dieser Informationen ergeben. Alle Informationen in diesem Artikel werden „wie besehen“ bereitgestellt, ohne Gewähr für Vollständigkeit, Richtigkeit, Aktualität oder die durch die Verwendung dieser Informationen erzielten Ergebnisse und ohne Gewährleistung jeglicher Art, weder ausdrücklich noch stillschweigend, einschließlich, aber nicht beschränkt auf Garantien der Leistung, Marktgängigkeit und Eignung für einen bestimmten Zweck. Nichts hierin ersetzt in irgendeiner Weise die unabhängigen Untersuchungen und das fundierte technische und geschäftliche Urteilsvermögen des Lesers. In keinem Fall haften Veeda Clinical Research oder seine Partner, Mitarbeiter oder Vertreter gegenüber dem Leser oder anderen Personen für Entscheidungen oder Maßnahmen, die im Vertrauen auf die Informationen in diesem Artikel getroffen werden, oder für Folgeschäden, besondere oder ähnliche Schäden wenn auf die Möglichkeit solcher Schäden hingewiesen wird. Kein Teil dieser Veröffentlichung darf ohne vorherige schriftliche Genehmigung des Herausgebers reproduziert, in einem Abrufsystem gespeichert oder in irgendeiner Form oder mit irgendwelchen Mitteln, sei es mechanisch, elektronisch, durch Fotokopieren, Aufzeichnen oder auf andere Weise, übertragen werden.
Für Informationen kontaktieren Sie uns unter:
Veeda Clinical Research Private Limited
Vedant Complex, neben dem YMCA Club, SG Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat Indien.
Telefon: + 91-79-3001-3000
Fax: + 91-79-3001-3010
Email: info@veedacr.com