
Indien entwickelt sich zu einem Land mit enormem Potenzial, einen Beitrag zu nationalen und internationalen Plattformen für klinische Studien zu leisten.
Das Zentrale Organisation zur Kontrolle von Arzneimittelstandards (CDSCO) ist die nationale Regulierungsbehörde Indiens, die vom Drug Controller General of India (DCGI) verwaltet wird.1,2,3
Das DCGI ist für die Koordinierung der Inspektionen der Sponsoren, Produktionseinheiten und Trail-Standorte verantwortlich.3
Frühe Jahre in der klinischen Entwicklung
Im Jahr 2000 legte der Indian Council of Medical Research (ICMR) ethische Richtlinien für die Durchführung biomedizinischer Forschung an Menschen fest.4 Im Jahr 2005 wurde Anhang Y des Arzneimittel- und Kosmetikgesetzes von 1945 überarbeitet, um die indischen Regulierungsgesetze an international anerkannte Definitionen und Verfahren anzupassen.
Zu den Änderungen gehörten:
- Definieren Phase I zur Phase IV einer Studie
- Abgegrenzte Verantwortlichkeiten von Sponsor(en) und Prüfer(n)
- Optionen zur Aufzeichnung etwaiger Abweichungen oder Änderungen am genehmigten Studienprotokoll
Indien unterzeichnete 2005 außerdem das Abkommen über handelsbezogene Rechte an geistigem Eigentum (TRIPS), um die Aussicht auf die Durchführung weiterer klinischer Studien in Indien zu eröffnen.5
Abgesehen von der Harmonisierung der Regulierungsgesetze an internationale Standards entwickelte sich Indien schnell zu einem bevorzugten Ziel für klinische Studien, da es Folgendes bot:6
- Englischsprachige Fachkräfte im Gesundheitswesen
- Technische Fachkentnis
- Wachsende Wirtschaft
- Weltklasse-Technologie
- Große, vielfältige und behandlungsnaive Bevölkerung
Zurückgesetzt für klinische Studien
Trotz Änderungen in den Vorschriften machten sich viele multinationale Pharmaunternehmen die große Bevölkerung zunutze, die entweder über unzureichende Kenntnisse über klinische Studien verfügte oder Analphabeten war. Darüber hinaus erschwerte ein schlecht definiertes Gesundheitssystem die Überwachung unethischer Praktiken.
Dies führte dazu, dass klinische Studien mit wenig Aufsicht durchgeführt wurden und die Einwilligung des Patienten weder schriftlich noch als audiovisueller Inhalt aufgezeichnet wurde.
Den Patienten wurden Prüfpräparate oder -geräte verabreicht, ohne dass bekannte schwerwiegende Nebenwirkungen bekannt wurden, von denen einige zum Tod der Probanden führten. Darüber hinaus wurde kein unabhängiger Untersuchungsausschuss eingerichtet, um festzustellen, ob der Tod des Patienten mit dem Prüfprodukt oder -gerät zusammenhängt bzw. nicht.4
Die Jahre 2010 bis 2013 waren geprägt von einer schwierigen Phase Indische klinische Studie Szenario aufgrund der akkumulierten negativen Auswirkungen der Durchführung unethischer Prozesse.
Allerdings verzeichnet das Clinical Trial Registry of India (CTRI) mit einem besseren Regulierungsrahmen einen stetigen Anstieg der Anzahl der durchgeführten Studien, wie in Abbildung 1 dargestellt. Es wurde auch beobachtet, dass es sich bei den meisten Studien um Phase III handelte Versuche.7
Abbildung 1: Trends bei klinischen Studien im Laufe der Jahre
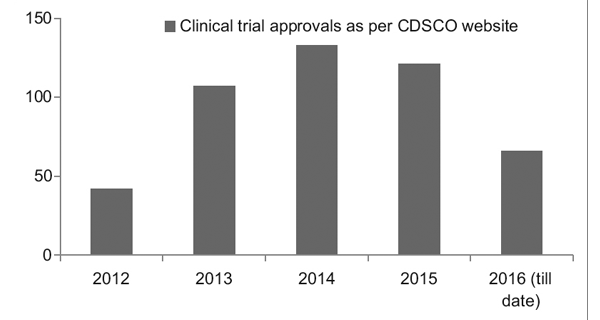
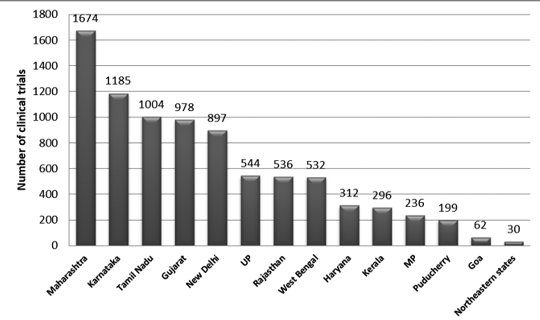
Abbildung 2 zeigt die bundesstaatliche Verteilung der Versuche in Indien zwischen 2007 und 2015. In diesem Zeitraum wurden etwa 3330 Versuche registriert.
Es wurde festgestellt, dass die höchste Anzahl an Gerichtsverfahren in Maharashtra und die geringste Anzahl an Gerichtsverfahren im nordöstlichen Bundesstaat durchgeführt wurde. In den nordöstlichen Bundesstaaten wurden in Nagaland keine Prozesse durchgeführt.7
Abbildung 2: Verteilung klinischer Studien in Indien nach Bundesstaaten (Daten 2007–2015)7
Wiederbelebung des klinischen und regulatorischen Szenarios
Im Jahr 2014 setzte die CDSCO 12 neue Drogenberatungsausschüsse (NDAC) und 25 Fachexpertenausschüsse (SECs) ein. Diese Ausschüsse verfügen über eine Reihe von Experten renommierter staatlicher Hochschulen und Institutionen, um die Genehmigungsfristen einer klinischen Studie auf 6 bis 7 Monate zu verkürzen.
Der dreistufige Prozess besteht aus:9
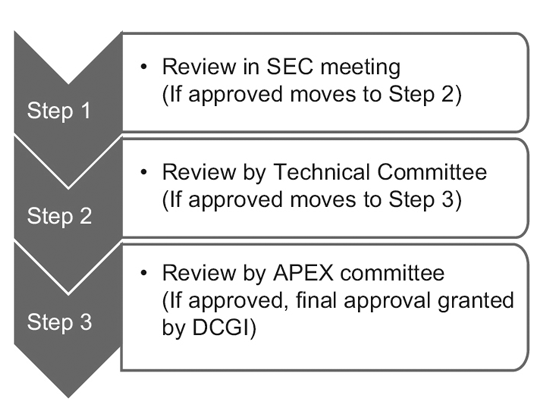
Allerdings prüft nur die SEC globale Anträge für klinische Studien und es ist keine weitere Genehmigung des Technischen Ausschusses oder des Apex-Ausschusses erforderlich. Anträge auf neue Prüfpräparate (IND) werden ebenfalls unabhängig vom IND-Ausschuss geprüft und bedürfen nicht der Genehmigung des Apex-Ausschusses.
Ein Technisches Komitee kommt nur dann ins Spiel, wenn die SEC den Antrag eines Sponsors abgelehnt hat und der Sponsor sich durch die Entscheidung beleidigt fühlt. Wenn das Technische Komitee in einem solchen Fall mit der Entscheidung der SEC nicht einverstanden ist, hat es die Befugnis, die Entscheidung der SEC außer Kraft zu setzen.10
Im März 2019 veröffentlichte das indische Ministerium für Gesundheit und Familienfürsorge die Neue Arzneimittel und Regeln für klinische Studien 2019 mit der Absicht, die Genehmigung für klinische Studien, neue Arzneimittel, Bioäquivalenzstudien (BE) oder Bioverfügbarkeitsstudien (BA) zu beschleunigen.
Mit diesen Regeln wurde auch etwaigen Unklarheiten Rechnung getragen, die im Hinblick auf die Regelungen der Ethikkommission (EK) bestanden.11
Höhepunkte der neuen Arzneimittel- und klinischen Studienregeln, 201911 |
Aktualisierte Regeln und Vorschriften11 |
Genehmigungszeitplan für klinische Studien |
90 Arbeitstage ab Eingang eines Antrags für Arzneimittel, die außerhalb Indiens entdeckt wurden, und 30 Arbeitstage für neue Arzneimittel oder IND in Indien |
Herstellung neuer Arzneimittel oder IND-, BE- und BA-Studien oder Testanalysen oder Untersuchungen |
Eine Genehmigung der Central Licensing Authority (CLA) ist erforderlich. |
Verzicht auf lokale klinische Studien |
· Wenn CLA die Vermarktung des neuen Medikaments in anderen Ländern genehmigt oder die Erlaubnis zur Durchführung globaler klinischer Studien für das neue Medikament in Indien erteilt hat · Keine Hinweise auf einen Unterschied im Stoffwechsel, in der Sicherheit oder in der Wirksamkeit aufgrund des unterschiedlichen genetischen Profils der indischen Bevölkerung |
Gültigkeitsdauer einer klinischen Studie |
2 Jahre ab Ausstellungsdatum durch CLA |
Zugang zu IND oder einem neuen Medikament nach dem Prozess |
Unter besonderen Umständen muss das Medikament gemäß den Anweisungen des CLA kostenlos an die Versuchspersonen verteilt werden, der Sponsor übernimmt jedoch keine Haftung für die Verwendung des Medikaments nach der Studie. |
Treffen vor und nach der Einreichung |
Um Rat bezüglich der Gesetze und Verfahren einzuholen, die den Prozess der Herstellung und Lizenzierung oder Erteilung von Genehmigungen regeln. |
Genehmigung für von EC durchgeführte Versuche und Registrierung von EC |
· Die Genehmigung muss von der EG eines anderen Versuchsstandorts eingeholt werden, wenn ein Versuchsstandort über keinen EG verfügt und der EG nicht weiter als 50 km vom Versuchsstandort entfernt sein sollte.· Eine vom CLA genehmigte EG-Registrierung bleibt ab dem Ausstellungsdatum fünf Jahre lang gültig. |
Bedingungen, die für die Durchführung einer klinischen Prüfung erfüllt sein müssen |
· Übermittlung eines Statusberichts vierteljährlich oder abhängig von der Dauer des Versuchs, um die Rekrutierung von Probanden zu verfolgen· Online-Bericht über den Status der klinischen Studie alle sechs Monate über das SUGHAM-Portal, um zu erfahren, ob die Studie noch läuft, abgeschlossen ist oder abgebrochen wurde. |
Gebühr für den Erwerb einer Lizenz, einer Registrierungsbescheinigung und einer Probeerlaubnis |
Unterschiedliche Gebührenstrukturen je nach Zweck des Versuchs. Gebühr zwischen 50,000 und 5,00,000 INR. |
Bridging the Gap
Die Herausforderungen im Umgang mit klinischen Studien sind vielfältig und erfordern die verantwortungsvolle und ethische Einhaltung des Regulierungsrahmens durch alle Beteiligten, die Regierung und das Justizsystem gleichermaßen.
Patientensicherheit und -schutz Es sollte von größter Bedeutung sein, strenge Regeln festzulegen für:12
- Einverständniserklärung durch audiovisuelle Aufzeichnung und in einer Sprache, mit der der Patient vertraut ist
- Respekt vor dem kulturellen, sozialen, wirtschaftlichen und pädagogischen Hintergrund des Patienten
- rechtzeitige Meldung von SAEs
Neue Grundregeln die die Möglichkeit eröffnen können, die medizinische Forschung in Indien auszuweiten, sind:13
- Genehmigung von Vorschlägen, die dem DCGI innerhalb von 30 Tagen nach Antragstellung vorgelegt werden, sofern keine Mitteilung des DCGI vorliegt
- schnelle Verfolgung inländischer Genehmigungen
- Treffen vor und nach der Einreichung mit dem Expertenausschuss, um mehr Transparenz in den Prozess zu bringen und einen genau definierten Zeitplan für den Abschluss des Versuchs festzulegen
- Studienentschädigung für den Fall, dass das Prüfpräparat zu schweren unerwünschten Ereignissen/Todesfällen führte.
Kompetente Arbeitskräfte und hochmoderne Infrastruktur spielen auch eine wichtige Rolle bei der Gewinnung von Sponsorunternehmen. Untersuchungen haben gezeigt, dass Phase-III-Studien zwar in großem Umfang in Indien durchgeführt werden, Phase-I-Studien jedoch auf das Sponsorland beschränkt zu sein scheinen.
Dies könnte auf die Bedenken des Sponsors bei der Beschaffung qualifizierter Arbeitskräfte und Technologie zurückzuführen sein. Damit einheimische Forschung in Indien stattfinden kann, ist es von entscheidender Bedeutung, dem Personal eine angemessene Exposition oder kontinuierliche medizinische Ausbildung sowie Zugang zu modernster Technologie zu bieten, um als Land anerkannt zu werden, das kompetent genug ist, alle Phasenversuche durchzuführen.9
Ebenso wichtig ist die Notwendigkeit, im ganzen Land qualifiziertes Gesundheitspersonal zur Verfügung zu haben, um der ungleichen Verteilung klinischer Studien in den einzelnen Bundesstaaten Rechnung zu tragen.
Die Konzentration einer Studie auf einen bestimmten Zustand könnte zu voreingenommenen Schlussfolgerungen führen und eine Krankheitslast oder einen Krankheitszustand zu stark vereinfachen oder übertreiben. Indem wir Menschen in allen Bundesstaaten den Zugang zur Teilnahme an einer klinischen Studie ermöglichen, minimieren wir nicht nur Vorurteile, sondern beziehen auch unterschiedliche ethnische Bevölkerungsgruppen ein.9
Die Zukunft
Mit positiven, patientenfreundlichen, schnellen und transparenten Regulierungsgesetzen wird Indien als internationales Zentrum für die Prüfung und Entwicklung innovativer Medikamente und medizinischer Geräte weiter wachsen.
Quellen
1. Evangeline L, Mounica NVN, Reddy VS et al. Regulierungsprozess und Ethik für klinische Studien in Indien (CDSCO). Das Pharma Innovation Journal. 2017;6(4):165-9. http://www.thepharmajournal.com/archives/2017/vol6issue4/PartC/6-4-4-176.pdf
2. Lahiry S., Sinha R., Choudhary S et al. Paradigmenwechsel in den Vorschriften für klinische Studien in Indien. Indisches Journal für Rheumatologie. 2018; 13: 51-5.
3. Gogtay NJ, Ravi R und Thatte UM. Regulatorische Anforderungen für klinische Studien in Indien: Was Akademiker wissen müssen. Indian Journal für Anästhesie. 2017 Mar;61(3):192-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5372399/
4. Ramu B, Kumar MS und Ramakrishna N. Aktuelles Regulierungsszenario für die Durchführung klinischer Studien in Indien. Pharmazeutische Regulierungsangelegenheiten. Offener Zugang. 2015, 4: 2. https://www.researchgate.net/publication/281765214_Current_Regulatory_Scenario_for_Conducting_Clinical_Trials_in_India
5. Burt T., Sharma P., Dhillon S et al. Klinisches Forschungsumfeld in Indien: Herausforderungen und Lösungsvorschläge. Journal of Clinical Research Bioethics. 2014;5:6. DOI: 10.4172/2155-9627.1000201
6. Chaturvedi M, Gogtay NJ, Thatte UM. Entsprechen die in Indien durchgeführten klinischen Studien den Gesundheitsbedürfnissen des Landes? Eine Prüfung des Clinical Trials Registry of India. Perspektiven in der klinischen Forschung. 2017;8(4):172-5.
7. http://ctri.nic.in/Clinicaltrials/news/CTRI_Newsbulletin_July-Dec_2017.pdf Zugriff am 23. April 2019.
8. Bhave A und Menon S. Regulatorisches Umfeld für die klinische Forschung: Jüngste Vergangenheit und erwartete Zukunft. Perspektiven in der klinischen Forschung. 2017, 8: 11.6.
9. Wichtige Highlights der neuen Arzneimittel und Regeln für klinische Studien, 2019. Zugriff am 23. April 2019
10. Dan S., Karmakar S., Ghosh B et al. Digitalisierung klinischer Studien in Indien: Ein neuer Schritt von CDSCO zur Gewährleistung der Glaubwürdigkeit der Daten und der Patientensicherheit. Pharmazeutische Zulassungsangelegenheiten: Open Access. 2015;4(3): DOI: 10.4172/2167-7689.1000149.
11 https://www.thehindubusinessline.com/news/new-rules-sweeten-the-deal-for-clinical-trials-by-indian-pharma-cos/article26283499.ece Zugriff am 23.
Haftungsausschluss:
Die in diesem Artikel enthaltenen Informationen dienen ausschließlich der allgemeinen Orientierung zu Themen, die für den persönlichen Gebrauch des Lesers von Interesse sind, der die volle Verantwortung für deren Verwendung übernimmt. Dementsprechend werden die Informationen in diesem Artikel mit der Maßgabe bereitgestellt, dass der/die Autor(en) und der/die Herausgeber hierin nicht mit der Erbringung professioneller Ratschläge oder Dienstleistungen befasst sind.
Daher sollte es nicht als Ersatz für die Beratung durch einen kompetenten Berater dienen. Bevor der Leser eine Entscheidung trifft oder Maßnahmen ergreift, sollte er immer einen professionellen Berater zu Rate ziehen, der sich auf den betreffenden Artikelbeitrag bezieht.
Obwohl alle Anstrengungen unternommen wurden, um sicherzustellen, dass die in diesem Artikel enthaltenen Informationen aus zuverlässigen Quellen stammen, ist Veeda Clinical Research nicht für etwaige Fehler oder Auslassungen oder für die Ergebnisse verantwortlich, die sich aus der Verwendung dieser Informationen ergeben.
Alle Informationen in diesem Artikel werden „wie besehen“ bereitgestellt, ohne Gewähr für Vollständigkeit, Richtigkeit, Aktualität oder die durch die Verwendung dieser Informationen erzielten Ergebnisse und ohne Gewährleistung jeglicher Art, weder ausdrücklich noch stillschweigend, einschließlich, aber nicht beschränkt auf Garantien der Leistung, Marktgängigkeit und Eignung für einen bestimmten Zweck.
Nichts hierin ersetzt in irgendeiner Weise die unabhängigen Untersuchungen und das gesunde technische und geschäftliche Urteilsvermögen des Lesers. Auf keinen Fall Klinische Veeda-Forschungoder seine Partner, Mitarbeiter oder Vertreter haften gegenüber dem Leser oder anderen Personen für Entscheidungen oder Maßnahmen, die im Vertrauen auf die Informationen in diesem Artikel getroffen werden, oder für Folgeschäden, besondere oder ähnliche Schäden, selbst wenn auf die Möglichkeit hingewiesen wurde solcher Schäden.
Kein Teil dieser Veröffentlichung darf ohne die vorherige schriftliche Genehmigung des Herausgebers reproduziert, in einem Abrufsystem gespeichert oder in irgendeiner Form oder mit irgendwelchen Mitteln, sei es mechanisch, elektronisch, durch Fotokopieren, Aufzeichnen oder auf andere Weise, übertragen werden.
Für weitere Informationen kontaktieren Sie uns unter:
Veeda Clinical Research Private Limited
Vedant Complex, neben dem YMCA Club, SG Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat Indien.
Telefon: + 91-79-3001-3000
Fax: + 91-79-3001-3010
Email: info@veedacr.com


{kind=link}