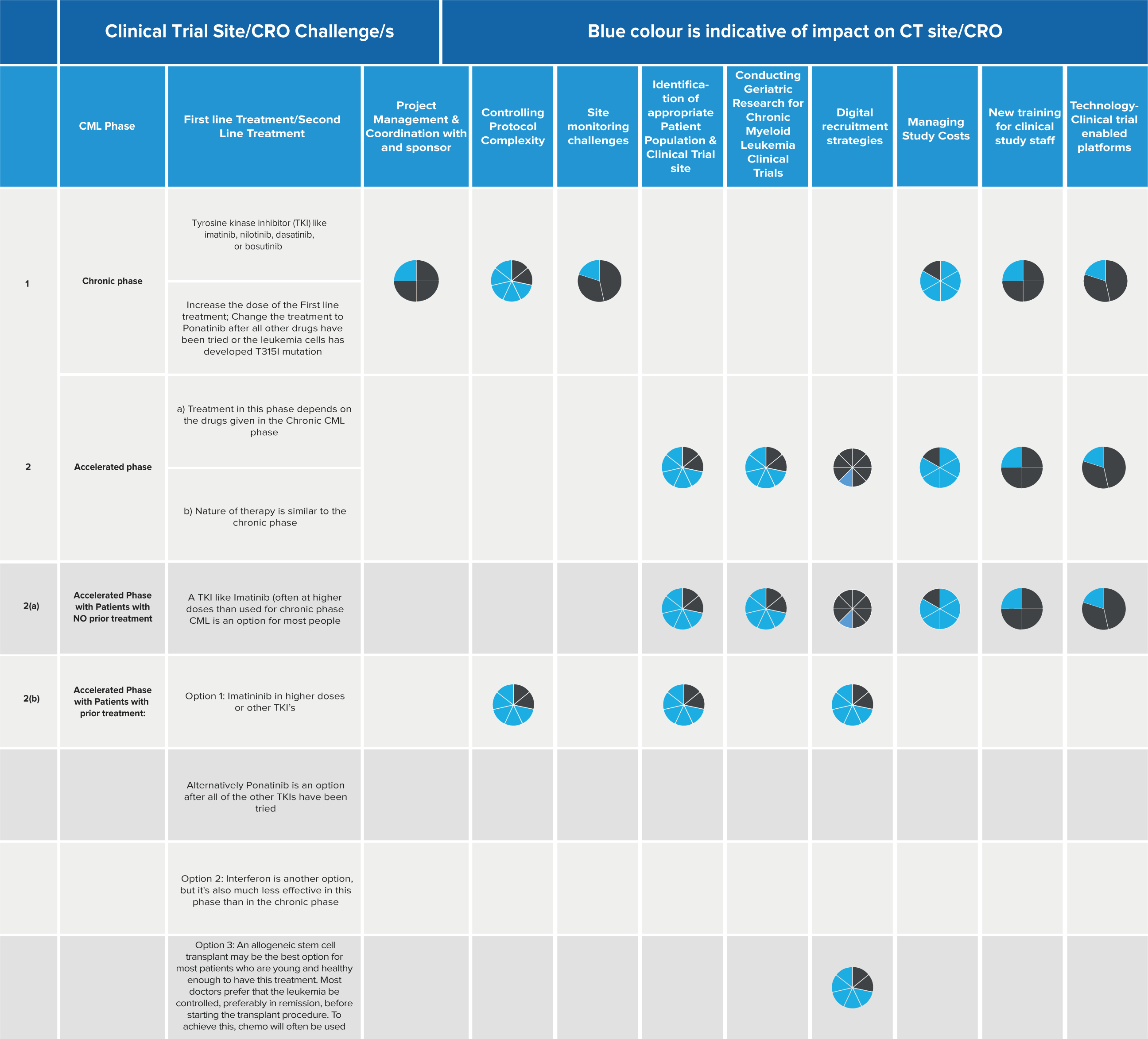
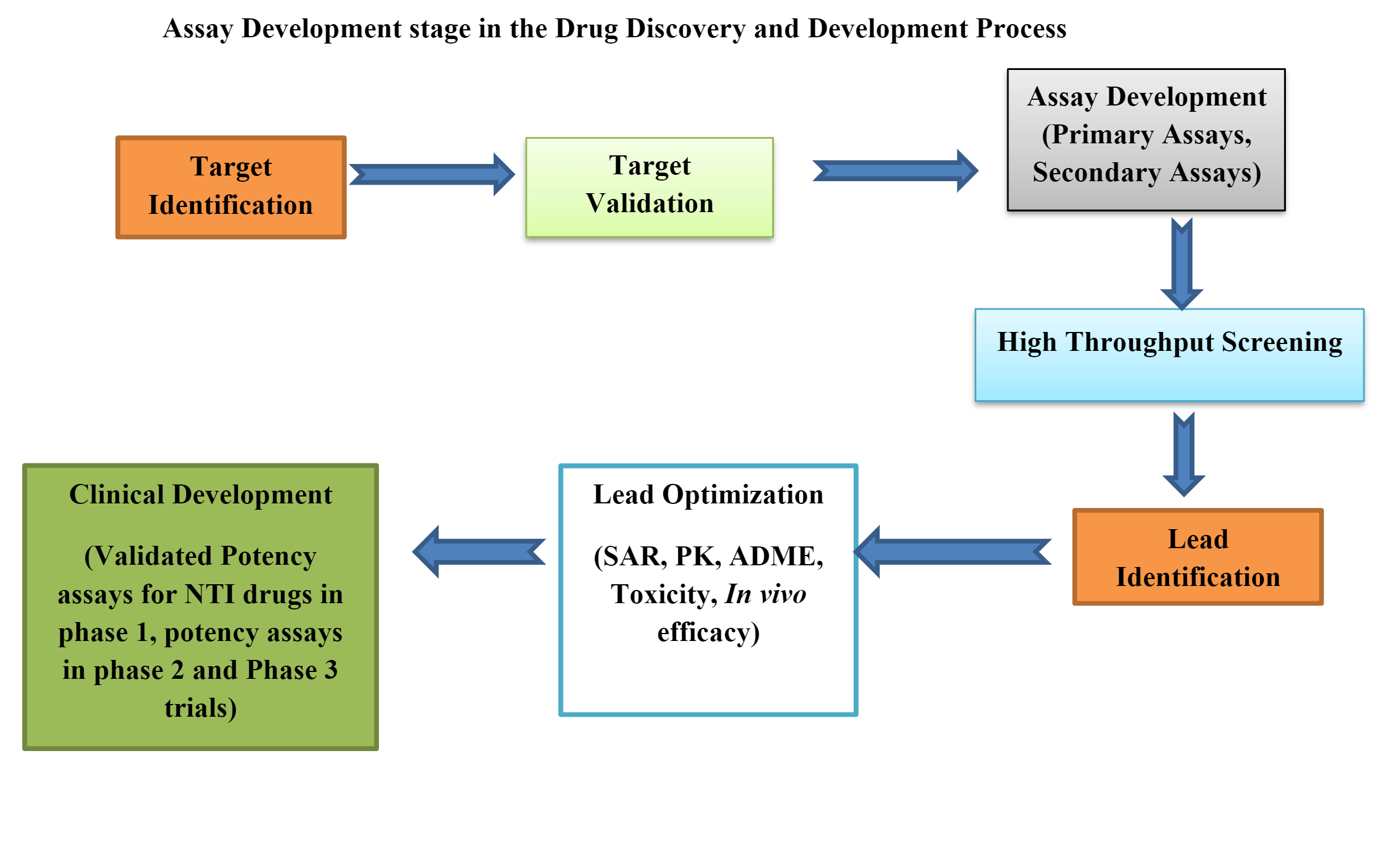
Bioanalytical labs play a crucial role in drug development, providing essential data to answer fundamental questions like “Does it work?” and “Is it safe?” The speed at which scientists can make informed decisions directly impacts the pace of bringing new drugs to market. To meet this challenge, labs are turning to digital solutions that streamline operations and improve data quality.
Unlocking the Power of Data
One of the key assets in modern bioanalytical labs is data. Smart data management can save time, reduce waste, and provide reliable answers quickly. However, in many labs, data is scattered across various systems, including paper notebooks, and spreadsheets. This fragmented approach makes it challenging to leverage data efficiently, leading to missed opportunities and inefficiencies.
With the rise of connected instruments and advanced analytical instruments like ELNs, LIMSs, labs can now integrate their data into a central backbone. This integration allows for streamlined operations, reduced human errors, and improved data accessibility. By centralizing data, labs can create user-friendly reports, and workflows, enabling scientists to make faster, more informed decision
Power of ELN, LIMS and LES for Bioanalysis
The digital transformation of bioanalytical labs is greatly facilitated by the use of Laboratory Information Management Systems (LIMS), Electronic Lab Notebooks (ELN), and Laboratory Execution Systems (LES). These systems play crucial roles in streamlining operations, improving data quality, and enhancing decision-making processes.
LIMS (Laboratory Information Management Systems):
LIMS are central to the implementation of a digital strategy in bioanalytical labs. They provide a structured framework for managing sample information throughout its lifecycle. By tracking sample details from login to disposition, LIMS ensure that data is captured accurately and consistently. This centralized approach to data management improves data integrity and accessibility, enabling scientists to make informed decisions more efficiently.
LIMS play a key role in integrating data from various sources, such as instruments, assays, and experiments. By providing a unified platform for data storage and management, LIMS enable labs to streamline operations and reduce manual errors. This integration also facilitates compliance with regulatory requirements, as data can be easily audited and traced back to its source.
Overall, LIMS contribute significantly to the efficiency and effectiveness of bioanalytical labs, enabling them to leverage data more effectively and make informed decisions.
ELN (Electronic Lab Notebooks):
ELNs are another essential tool in the digital transformation of bioanalytical labs. They provide a digital platform for recording and managing experimental data, replacing traditional paper lab notebooks. ELNs offer several advantages over paper notebooks, including the ability to standardize workflows, automate data entry, and facilitate collaboration among scientists.
One of the key benefits of ELNs is their ability to standardize experimental workflows. By providing templates for recording experimental details, ELNs ensure that data is captured consistently and accurately. This standardization not only improves data quality but also makes it easier to search and analyze data.
ELNs also facilitate collaboration among scientists by providing a central platform for sharing and accessing experimental data. This collaborative approach to data management enables scientists to work more efficiently and effectively, leading to faster decision-making and better outcomes.
LES (Laboratory Execution Systems):
LES are specialized systems designed to automate and enforce procedural steps in the laboratory. In the context of bioanalytical labs, LES play a crucial role in ensuring that experiments are conducted consistently and according to standard operating procedures (SOPs).
One of the key advantages of LES is their ability to enforce procedural execution during testing. By encapsulating SOPs into software, LES ensure that each step of the testing process is recorded and completed before moving on to the next step. This not only improves data quality but also reduces the risk of errors and deviations from protocol.
LES also facilitate real-time monitoring of experiments, allowing scientists to make informed decisions based on up-to-date data. This real-time feedback loop enables labs to respond quickly to changing conditions and optimize experimental workflows for better results
Refining Bioanalytical Labs: Unifying Digital Solutions for Efficiency, Quality, and Innovation
1. Deliver a Platform-Based yet Personalized Laboratory Experience
While personalization of laboratory technologies can be beneficial in the short term, it often leads to information silos and challenges in information exchange. A platform-based approach, on the other hand, allows labs to leverage integrated modules aligned with standard enterprise-wide R&D terminologies and capabilities. This approach, facilitated by tools like LIMS and ELNs, enables better-quality study data generation and enhances collaboration among researchers. By adopting harmonized approaches across sites, labs can achieve enhanced visibility, real-time tracking of experiment statuses, and improved cross-experimental insights.
2. Leverage Digital Lab Tools to Unlock Operational Efficiency & Cost Savings
Digital lab technologies such as LIMS, ELNs, and quality management systems offer significant operational efficiencies and cost-saving opportunities. By retiring legacy systems, eliminating redundant data entry, and building audit trails, labs can streamline workflows, ensure data accuracy, and enhance compliance with regulatory requirements. Additionally, these technologies reduce employee time spent on manual tasks and enable real-time tracking of project workloads, leading to substantial time savings per employee.
3. Drive Enhanced Data Reproducibility & Data Analysis to Create Commercial Value
Data reproducibility is a critical challenge in bioanalytical labs, leading to wasted time, decreased resources, and lower scientific output. Digital platforms that enhance data quality and increase statistical power can address this challenge. By standardizing higher-quality data, labs can increase reproducibility and improve experimental performance. Furthermore, leveraging data analytics tools can help labs extract additional value from their data, accelerating the discovery of new indications and molecules.
Veeda’s Integration of LIMS, ELN, and LES Solutions
Veeda’s Bioanalysis solution integrates Laboratory Information Management System (LIMS), Electronic Laboratory Notebook (ELN), and Laboratory Execution System (LES) functionalities to optimize our bioanalytical lab operations. This integrated approach for bioanalytical studies by providing advanced data management, analysis, and automation tools in a single, cohesive system.
LIMS centralizes sample tracking and data management, ensuring traceability and compliance with regulatory standards. Meanwhile, ELN digitizes experimental data, improving collaboration and reducing manual errors. The LES further enhances our workflows by automating processes and enforcing SOPs, ensuring consistency and quality in our operations. This integration enhances our bioanalytical procedures into efficient, reliable testing methods, where we leveraging connected instruments and intelligent data management capabilities to consistently improve our deliverable outcomes.
Reference Articles:
https://www.technologynetworks.com/informatics/articles/eln-lims-cds-les-whats-the-difference-313834
https://www.labware.com/blog/streamlining-bioanalytical-testing-with-a-unified-lims-and-eln-solution