Bioanalyser är inblandade i varje steg av läkemedelsupptäckten, från målidentifiering tills man upptäcker huvudföreningen. Bioanalyser ger värdefull information som visar den terapeutiska styrkan hos ett läkemedel som undersöks.
Data som genereras under bioassay spelar också en viktig roll i läkemedelsutveckling och kvalitetskontroll av färdiga biologiska produkter. Rätt utformade bioanalyser hjälper till att bedöma den biologiska effekten, aktiviteten, signaltransduktionsprocessen och receptorbindningsförmågan hos läkemedelsprodukt eller biologiskt ämne på ett biologiskt mål (proteiner) jämfört med en referens eller standard över ett lämpligt biologiskt system.
De läkemedels- och bioteknikföretag som är involverade i läkemedelsupptäckt och utveckling utmanas kontinuerligt med att utveckla biologiskt relevanta analyser för analys av flera potentiella mekanismer.
Processen involverar användning av kvalitetskritiska reagenser, användning av specifika cellinjer och renade testläkemedel och referensläkemedelsprodukter som ibland kan bli en begränsning. De flesta av dessa aktiviteter kräver tillräckligt med tid, vilket kan bli en begränsande faktor för biofarmakatillverkare.
Det är värt att lägga ut aktiviteter på entreprenad välrenommerade CRO-tjänsteleverantörer för att spara tid i utvecklingsinsatser och även att ha en opartisk åsikt om läkemedlets funktionella aktiviteter.
Veeda Group har kvalificerade och erfarna forskare för att designa, utveckla, utföra och validera bioanalyserna för företag och tillhandahåller förstklassiga bioanalystjänster (vitro och in vivo-) som genererar meningsfull data för att stödja läkemedels- och bioteknikföretag i deras läkemedelsupptäckts- och utvecklingsresa.
Veeda Groups erfarenhet av utveckling och genomförande av bioanalyser inkluderar:
- Plackreduktionsneutraliseringstest (PRNT-analys)
- vitro Hudsensibilisering Aktiveringstest för mänsklig celllinje (h-CLAT-analys)
- Nab-analys
- Analysutveckling (farmakodynamik, farmakokinetik, immunogenicitet och bedömning av biomarkörer)
- In vivo Bioanalyser för läkemedelsmolekyler som luteiniserande hormon, epoetin, HCG, rekombinant FSH, β-HCG och insulin.
- ADCC-analys för biosimilarer och olika andra analyser som Ex Vivo analys, cellbaserad analys, receptorbindningsanalys, cytokinfrisättningsanalys och ADA-analys.
Veeda Group tillhandahåller integrerade upptäckts-, utvecklings- och regleringstjänster med sina flera teknikplattformar:
- Exploratoriska toxikologiska studier
- Regulatoriska toxikologiska studier
- In vitro Bioanalyser
- Ex vivo Bioanalyser
Gruppen har också erfarenhet av att hantera ett brett spektrum av bioterapeutika som terapeutiska monoklonala antikroppar, insulin- och insulinanaloger, cytokiner, hepariner med låg molekylvikt, biosimilars, Hormoner och biomarkörer.
Veeda-gruppen har visat förmåga att utveckla rekombinanta proteiner såsom icke-glykosylerade proteiner och glykoproteiner härledda från antingen bakteriella eller däggdjursvärduttryckssystem.
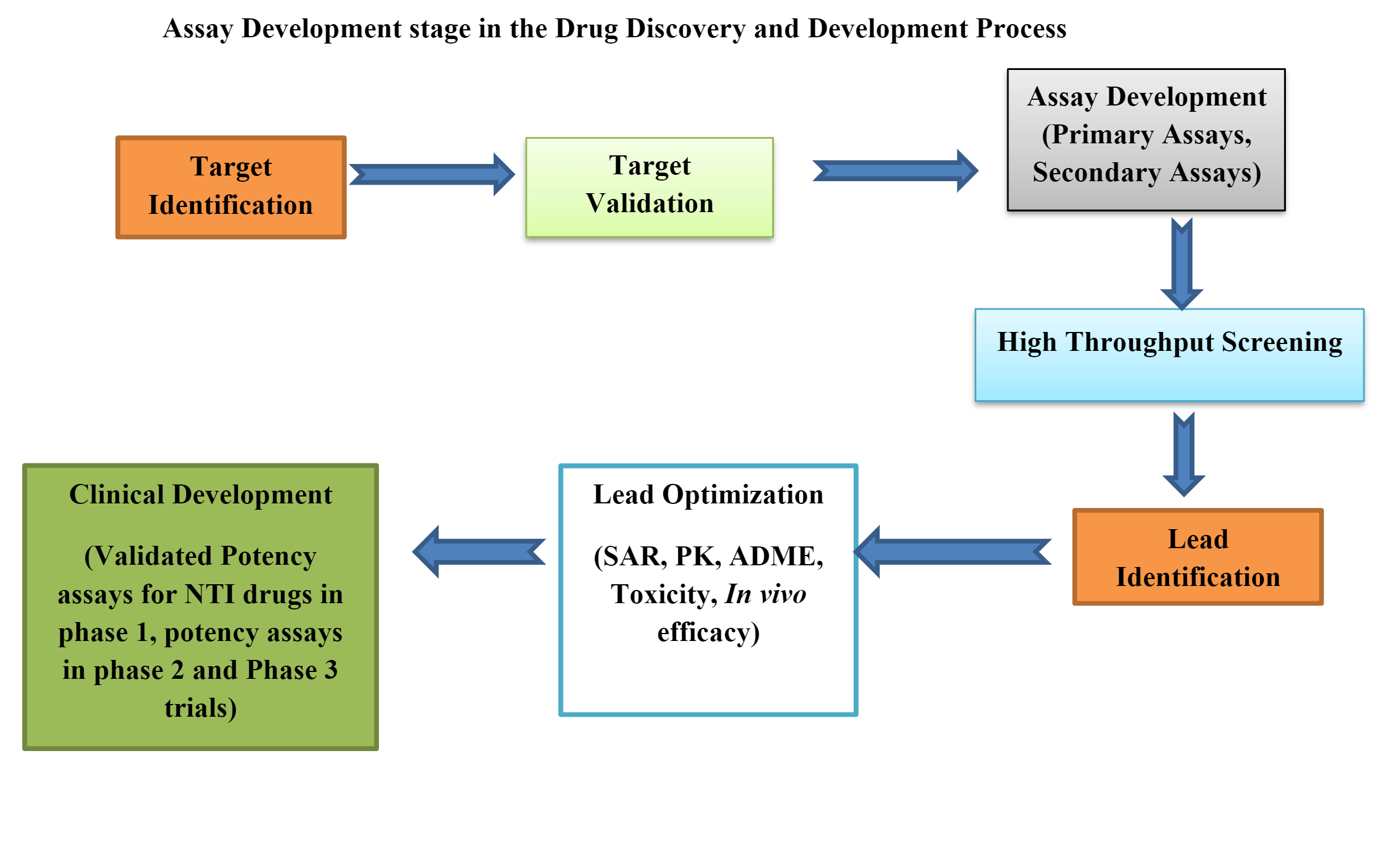
Bioanalyser i preklinisk läkemedelsutveckling
Biologiska analyser eller bioanalyser är viktiga verktyg i preklinisk läkemedelsutveckling. Prekliniska bioanalyser kan vara in vivo, ex vivooch vitro.
In vivo bioanalyser ger ett mer realistiskt och förutsägande mått på de funktionella effekterna av tester med referensläkemedelsprodukter eller standardmaterial med definierad styrka, tillsammans med tillämpning av statistiska verktyg, studiespecifika labbtekniker och efterlevnad av det väldesignade studieprotokollet.
Dessa analyser fångar komplexiteten i målengagemang, metabolism och farmakokinetik hos nya läkemedel bättre än vitro bioanalyser.
De mest använda experimentella däggdjuren i bl.an vivo effektivitetsanalyser är möss och råttor. Ibland kan andra arter användas beroende på analysernas känslighet och lämplighet.
Utveckling och validering av bioanalyser
Bioanalyser används som en screeningsmetod för att identifiera de signaler som indikerar önskad biologisk aktivitet från en uppsättning föreningar. I allmänhet kan två olika typer av signaler genereras av en bioanalys, en linjär dosrespons och en sigmoidal (S-formad) dosrespons.
Eftersom en lösning inte passar alla bioanalyser är det bra att utvärdera och analysera data för att utveckla en exakt metod för att utföra varje bioanalys.
Livscykelstadierna för en bioanalys är indelade i:
Steg 1: Metoddesign, utveckling och optimering
Steg 2: Procedurprestandakvalificering
Steg 3: Procedurprestandaverifiering (passar för ändamål)
Att ta fram en bioanalys som uppfyller regulatoriska krav och få en läkemedelsprodukt registrerad är en mycket komplex process.
Att utveckla en bioassay inkluderar många strategier och taktiska konstruktioner som att välja rätt in vivo- plattform, korrekt metod eller plattdesign, dataanalys, system/prov hållbarhetsstrategi, metodimplementering, metodprestanda och övervakning.
Det finns flera steg som ska följas för utveckling och validering av bioanalyser, såsom dos-respons och val av kurvanpassning, utveckling av referens, beräkning av styrka, bioassaykarakterisering, design av bioassay-kalkylator, standardisering och automatisering av bioassay, och slutligen , utvärdering.
Både metodutveckling och validering av bioanalyser inkluderar tre grundläggande områden:
- Validering av förstudie (identifikation och designfas).
- In-studie (Utvecklings- och produktionsfas) validering
- Korsvalidering eller metodöverföringsvalidering
Under metodutvecklingen väljs analysförhållanden och procedurer som minimerar inverkan av potentiella invaliditetskällor. Kommer till den statistiska valideringen för en in vivo- analys, den involverar fyra huvudkomponenter:
- Adekvat studiedesign och dataanalysmetod
- Korrekt randomisering av djur
- Lämplig statistisk kraft och urvalsstorlek
- Tillräcklig reproducerbarhet över analyskörningar.
Parallell gruppdesign, randomiserad blockdesign, upprepade åtgärder design och crossover design är de grundläggande typerna av experimentell design som används i in vivo- analysera.
Följande är nyckelfaktorerna som bör tänkas på när du utformar en in vivo- analysera:
- Alla betydelsefulla biologiska effekter (farmakologiskt) bör vara statistiskt signifikanta.
- Om biologiskt relevanta analyser inte finns, kan en rad rimliga effekter övervägas.
- De viktigaste effektmåtten bör vara väldefinierade innan analysen påbörjas.
- Djuren bör fördelas slumpmässigt på lämpligt sätt till behandlingsgrupperna.
- Dosnivåerna bör väljas på lämpligt sätt. Val av dos och kurvanpassning är bland de mest kritiska aspekterna av utvecklingen av bioanalys. Dosen bestäms beroende på vilken typ av modell som används i signalen för att passa data. För Sigmoidal design passar en logistikmodell med fyra eller fem parametrar (4PL eller 5PL) data, medan för linjär design passar en parallell linjeanalys (PLA) modell för data.
För en 4PL-modell rekommenderas nio doser:
- Tre doser i den nedre asymptoten
- Tre doser i den övre asymptoten
- Tre doser i det linjära området
För en PLA-modell rekommenderas däremot minst fyra doser. Minst tre på varandra följande doser krävs för att plotta doskurvan.
- Valet av kontrollgrupper och tidpunkter för provtagning bör vara optimalt.
- Designstrategierna bör minimera variationen och maximera informationen.
Att förstå design, utveckling och statistisk validering av in vivo- bioassay mer detaljerat, kontakta oss på https://www.veedacr.com. Man kan också läsa riktlinjerna som nämns av NIH genom att besöka länken:
https://www.ncbi.nlm.nih.gov/books/NBK92013/pdf/Bookshelf_NBK92013.pdf
Referensprojekt
- A. Little, "Essentials in Bioassay Development", BioPharm International 32 (11) 2019
- Padmalayam, Ph.D., Analysutveckling inom läkemedelsupptäckt
- Zwierzyna M, Overington JP (2017) Klassificering och analys av en stor samling in vivo bioanalysbeskrivningar. PLoS Comput Biol13(7): e1005641. https://doi.org/10.1371/journal.pcbi.1005641
- White JR, Abodeely M, Ahmed S, Debauve G, Johnson E, Meyer DM, Mozier NM, Naumer M, Pepe A, Qahwash I, Rocnik E, Smith JG, Stokes ES, Talbot JJ, Wong PY. Bästa praxis för utveckling av bioanalyser för att stödja registrering av bioläkemedel. Biotekniker. 2019 sep;67(3):126-137. doi: 10.2144/btn-2019-0031. Epub 2019 5 augusti. PMID: 31379198.
- F Chana och Hursh D, Bioassays through the Product Lifecycle: Perspectives of CDER and CBER reviews.
- Haas J, Manro J, Shannon H, et al. In vivo analysriktlinjer. 2012 maj 1 [Uppdaterad 2012 oktober 1]. I: Markossian S, Grossman A, Brimacombe K, et al., redaktörer. Analysvägledning [Internet]. Bethesda (MD): Eli Lilly & Company och National Center for Advancing Translational Sciences; 2004-. Bokhylla URL: https://www.ncbi.nlm.nih.gov/books/