
L’Inde émerge comme un pays doté d’un énorme potentiel pour contribuer aux plateformes nationales et internationales d’essais cliniques.
Les Organisation centrale de contrôle des normes pharmaceutiques (CDSCO) est l'autorité nationale de réglementation de l'Inde, gérée par le Drug Controller General of India (DCGI).1,2,3
La DCGI est responsable de la coordination des inspections des commanditaires, des unités de fabrication ainsi que des sites de sentiers.3
Premières années de développement clinique
En 2000, le Conseil indien de la recherche médicale (ICMR) a établi des lignes directrices éthiques pour mener des recherches biomédicales sur des sujets humains.4 L'année 2005 a vu une révision de l'annexe Y de la loi sur les médicaments et les cosmétiques de 1945, afin d'aligner les lois réglementaires indiennes sur les définitions et les procédures acceptées au niveau international.
Les changements comprenaient :
- Définir Phase I à la phase IV d'un essai
- Responsabilités délimitées du (des) promoteur (s) et du (des) chercheur (s)
- Options pour enregistrer tout écart ou modification du protocole d'étude approuvé
L'Inde a également signé l'accord sur les droits de propriété intellectuelle qui touchent au commerce (ADPIC) en 2005 afin d'ouvrir la possibilité de mener davantage d'essais cliniques en Inde.5
Outre l’harmonisation des actes réglementaires avec les normes internationales, l’Inde est rapidement devenue une destination favorable pour les essais cliniques car elle offrait :6
- Professionnels anglophones du secteur de la santé
- Expertise technique
- Une économie en croissance
- Technologie de classe mondiale
- Population nombreuse, diversifiée et naïve de traitement
Recul pour les essais cliniques
Malgré les changements apportés à la réglementation, de nombreuses sociétés pharmaceutiques multinationales ont profité d’une population nombreuse qui soit avait des connaissances insuffisantes en matière d’essais cliniques, soit était analphabète. En outre, un système de santé mal défini a ajouté aux défis liés à la surveillance des pratiques contraires à l’éthique.
Cela a conduit à mener des essais cliniques avec peu de supervision et sans enregistrement du consentement éclairé du patient, que ce soit sous forme écrite ou sous forme de contenu audiovisuel.
Les patients ont reçu des médicaments ou des dispositifs expérimentaux sans révéler d’effets indésirables graves connus, certains entraînant la mort des sujets. De plus, aucune commission d’enquête indépendante n’a été mise en place pour déterminer si le décès du patient était lié ou non au produit ou dispositif expérimental.4
Les années 2010 à 2013 ont été marquées par une phase éprouvante dans le Essai clinique indien scénario en raison des effets néfastes accumulés de la conduite de procès contraires à l’éthique.
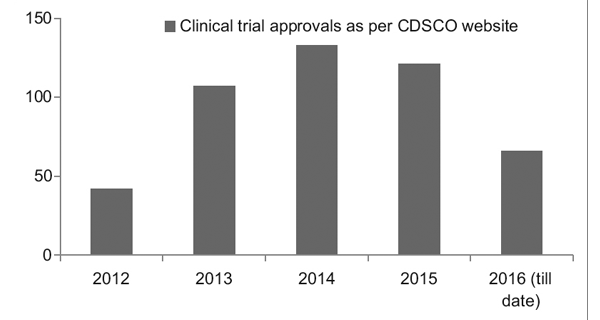
Cependant, grâce à un meilleur cadre réglementaire en place, le Clinical Trial Registry of India (CTRI) a enregistré une augmentation constante du nombre d'essais menés, comme le montre la figure 1. Il a également été observé que la plupart des essais étaient de phase III. essais.7
Figure 1: Tendances des essais cliniques au fil des ans
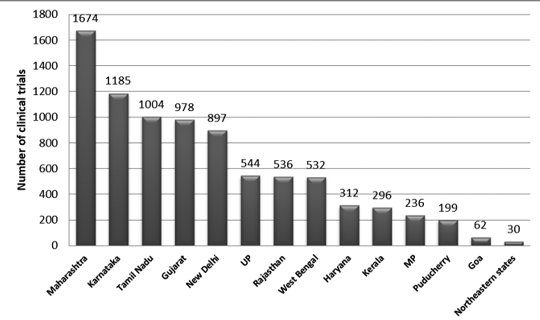
La figure 2 présente la répartition des essais par État en Inde entre 2007 et 2015. Environ 3330 XNUMX sentiers ont été enregistrés au cours de cette période.
Il a été observé que le nombre maximum d’essais a été mené dans le Maharashtra et que le nombre le plus faible d’essais a été mené dans l’État du Nord-Est. Parmi les États du Nord-Est, aucun essai n’a été mené au Nagaland.7
Figure 2: Répartition des essais cliniques par État en Inde (données 2007-2015)7
Reprise du scénario clinique et réglementaire
En 2014, le CDSCO a constitué 12 comités consultatifs sur les nouveaux médicaments (NDAC) et 25 comités d'experts en la matière (SEC). Ces comités comptent un certain nombre d'experts provenant d'éminents collèges et institutions gouvernementaux pour accélérer les délais d'approbation d'un essai clinique à 6 à 7 mois.
Le processus à trois niveaux comprend :9
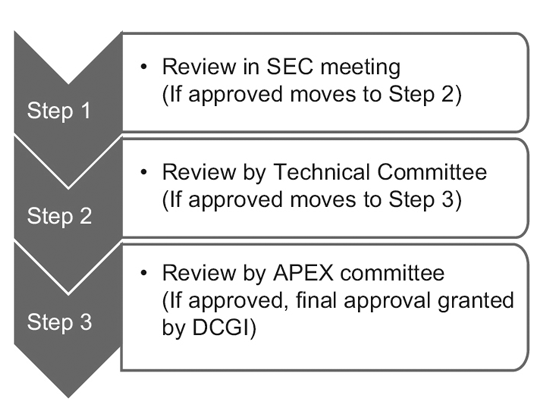
Cependant, seule la SEC examine les demandes d'essais cliniques mondiaux, et aucune autre approbation n'est requise de la part du comité technique ou du comité Apex. Les demandes de nouveaux médicaments expérimentaux (IND) sont également examinées de manière indépendante par le comité IND et ne nécessitent pas l'approbation du comité Apex.
Un comité technique n'intervient que si la SEC a rejeté la demande d'un sponsor et que celui-ci se sent lésé par la décision. Dans un tel cas, si le comité technique n'est pas d'accord avec la décision de la SEC, il a le pouvoir d'annuler la décision de la SEC.10
En mars 2019, le ministère indien de la Santé et du Bien-être familial a publié le Règles sur les nouveaux médicaments et les essais cliniques 2019 avec l'intention d'accélérer l'approbation des essais cliniques, des nouveaux médicaments, des études de bioéquivalence (BE) ou de biodisponibilité (BA).
Ces règles ont également levé toute ambiguïté qui existait en ce qui concerne la réglementation du Comité d'éthique (CE).11
Points saillants des règles sur les nouveaux médicaments et les essais cliniques, 201911 |
Règles et réglementations mises à jour11 |
Calendrier d’approbation pour les essais cliniques |
90 jours ouvrables à compter de la réception d'une demande pour les médicaments découverts en dehors de l'Inde et 30 jours ouvrables pour les nouveaux médicaments ou IND en Inde |
Fabrication de nouveaux médicaments ou études IND, BE & BA ou tests, analyses ou examens |
L'autorisation est requise auprès de la Central Licensing Authority (CLA) |
Renonciation aux essais cliniques locaux |
· Si la CLA a approuvé la commercialisation du nouveau médicament dans d'autres pays ou a accordé l'autorisation de mener des essais cliniques mondiaux pour le nouveau médicament en Inde. · Aucune preuve d'une différence de métabolisme, de sécurité ou d'efficacité en raison de la différence de profil génétique de la population indienne |
Durée de validité d'un essai clinique |
2 ans à compter de la date d'émission par CLA |
Accès après l'essai à l'IND ou à un nouveau médicament |
Dans des circonstances particulières, le médicament doit être distribué gratuitement aux sujets de l'essai selon les instructions de la CLA, mais aucune responsabilité n'incombe au promoteur quant à l'utilisation du médicament après l'essai. |
Réunions pré-soumission et post-soumission |
Demander des conseils sur la loi et les procédures qui régissent le processus de fabrication et d'octroi de licences ou d'autorisations. |
Approbation des essais menés par EC et enregistrement de EC |
· L'approbation doit être obtenue du CE d'un autre site d'essai si un site d'essai ne dispose pas d'un CE et que le CE doit se trouver à moins de 50 km du site d'essai.· L'enregistrement de EC approuvé par la CLA reste valable cinq ans à compter de la date d'émission. |
Conditions à remplir pour la conduite d’un essai clinique |
· Soumission d'un rapport de situation sur une base trimestrielle ou en fonction de la durée de l'essai pour suivre l'inscription des sujets· Rapport en ligne de l'état de l'essai clinique tous les six mois via le portail SUGHAM pour savoir si l'essai est en cours, terminé ou terminé. |
Frais pour l'obtention d'une licence, d'un certificat d'enregistrement et d'une autorisation d'essai |
Différentes structures tarifaires en fonction du but de l’essai. Frais allant de 50,000 5,00,000 à XNUMX XNUMX XNUMX INR. |
Combler le fossé
Les défis liés aux essais cliniques comportent de multiples facettes et impliquent le respect du cadre réglementaire de manière responsable et éthique par les parties prenantes, le gouvernement et le système judiciaire.
Sécurité et protection des patients Il devrait être de la plus haute importance d’établir des règles strictes pour :12
- consentement éclairé par enregistrement audiovisuel et dans une langue avec laquelle le patient est à l'aise
- le respect du milieu culturel, social, économique et éducatif du patient
- déclaration en temps opportun des EIG
De nouvelles règles de base qui peuvent ouvrir la possibilité d’étendre la recherche médicale en Inde sont :13
- approbation des propositions soumises à la DCGI dans les 30 jours suivant la demande, en l'absence de communication de la DCGI
- suivi rapide des approbations nationales
- réunions pré et post-soumission avec le comité d'experts pour apporter plus de transparence au processus et fixer un calendrier bien défini pour l'achèvement de l'essai
- indemnisation à l'essai au cas où le médicament expérimental entraînerait des EIG ou la mort.
Une main-d’œuvre compétente et une infrastructure de pointe jouent également un rôle important pour attirer des entreprises sponsors. Des recherches ont démontré que même si les essais de phase III sont menés à grande échelle en Inde, les essais de phase I semblent se limiter au pays sponsor.
Cela pourrait être attribué à l'appréhension du promoteur à se procurer une main-d'œuvre qualifiée et une technologie. Pour permettre à la recherche autochtone de se dérouler en Inde, il est essentiel de fournir une exposition appropriée ou une formation médicale continue au personnel et un accès à une technologie de pointe afin d'être reconnu comme un pays suffisamment compétent pour mener des essais de phase.9
Il est tout aussi important de disposer de professionnels de santé qualifiés dans tout le pays pour tenir compte de la répartition inégale des essais cliniques entre les États.
Concentrer un essai sur un état particulier pourrait conduire à des conclusions biaisées et simplifier à l’excès ou exagérer une charge de morbidité ou une affection. En permettant aux personnes de tous les États de participer à un essai clinique, nous minimisons non seulement les préjugés, mais incluons également diverses populations ethniques.9
El futuro
Grâce à des lois réglementaires positives, conviviales pour les patients, rapides et transparentes, l’Inde continuera de se développer en tant que plaque tournante internationale pour tester et développer des médicaments et des dispositifs médicaux innovants.
Sources
1. Évangéline L, Mounica NVN, Reddy VS et al. Processus réglementaire et éthique des essais cliniques en Inde (CDSCO). Le journal de l'innovation pharmaceutique. 2017;6(4):165-9. http://www.thepharmajournal.com/archives/2017/vol6issue4/PartC/6-4-4-176.pdf
2. Lahiry S, Sinha R, Choudhary S et al. Changement de paradigme dans la réglementation des essais cliniques en Inde. Journal indien de rhumatologie. 2018; 13: 51-5.
3. Gogtay NJ, Ravi R et Thatte UM. Exigences réglementaires pour les essais cliniques en Inde : ce que les universitaires doivent savoir. Journal indien d'anesthésie. 2017 Mar;61(3):192-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5372399/
4. Ramu B, Kumar MS et Ramakrishna N. Scénario réglementaire actuel pour la conduite d'essais cliniques en Inde. Affaires réglementaires pharmaceutiques. Accès libre. 2015; 4: 2. https://www.researchgate.net/publication/281765214_Current_Regulatory_Scenario_for_Conducting_Clinical_Trials_in_India
5. Burt T, Sharma P, Dhillon S et al. Environnement de recherche clinique en Inde : défis et solutions proposées. Journal de bioéthique de la recherche clinique. 2014;5:6. DOI : 10.4172/2155-9627.1000201
6. Chaturvedi M, Gogtay NJ, Thatte UM. Les essais cliniques menés en Inde correspondent-ils à ses besoins en matière de soins de santé ? Un audit du registre des essais cliniques de l'Inde. Perspectives en recherche clinique. 2017;8(4):172-5.
7. http://ctri.nic.in/Clinicaltrials/news/CTRI_Newsbulletin_July-Dec_2017.pdf Consulté le 23 avril 2019.
8. Bhave A et Menon S. Environnement réglementaire pour la recherche clinique : passé récent et avenir attendu. Perspectives en recherche clinique. 2017; 8: 11.6.
9. Points saillants des règles sur les nouveaux médicaments et les essais cliniques, 2019. Consulté le 23 avril 2019.
10. Dan S, Karmakar S, Ghosh B et al. Numérisation des essais cliniques en Inde : une nouvelle étape du CDSCO pour garantir la crédibilité des données et la sécurité des patients. Affaires réglementaires pharmaceutiques : libre accès. 2015 ; 4(3) : DOI : 10.4172/2167-7689.1000149.
11. https://www.thehindubusinessline.com/news/new-rules-sweeten-the-deal-for-clinical-trials-by-indian-pharma-cos/article26283499.ece Consulté le 23 avril 2019.
Avertissement:
Les informations contenues dans cet article sont destinées uniquement à fournir des orientations générales sur des questions d'intérêt pour l'usage personnel du lecteur, qui accepte l'entière responsabilité de son utilisation. En conséquence, les informations contenues dans cet article sont fournies étant entendu que les auteurs et les éditeurs ne sont pas engagés ici à fournir des conseils ou des services professionnels.
En tant que tel, il ne doit pas se substituer à la consultation d’un conseiller compétent. Avant de prendre une décision ou d'entreprendre une action, le lecteur doit toujours consulter un conseiller professionnel concernant la publication de l'article concerné.
Bien que tous les efforts aient été faits pour garantir que les informations contenues dans cet article proviennent de sources fiables, Veeda Clinical Research n'est pas responsable des erreurs ou omissions ou des résultats obtenus grâce à l'utilisation de ces informations.
Toutes les informations contenues dans cet article sont fournies « telles quelles », sans aucune garantie d'exhaustivité, d'exactitude, d'actualité ou des résultats obtenus grâce à l'utilisation de ces informations, et sans garantie d'aucune sorte, expresse ou implicite, y compris, mais sans s'y limiter. aux garanties de performance, de qualité marchande et d’adéquation à un usage particulier.
Rien dans le présent document ne saurait, dans aucune mesure, remplacer les enquêtes indépendantes et le bon jugement technique et commercial du lecteur. En aucun cas Recherche clinique Veeda, ou ses partenaires, employés ou agents, ne seront responsables envers le lecteur ou toute autre personne de toute décision ou action prise sur la base des informations contenues dans cet article ou de tout dommage consécutif, spécial ou similaire, même s'il est informé de la possibilité de tels dommages.
Aucune partie de cette publication ne peut être reproduite, stockée dans un système de récupération ou transmise sous quelque forme ou par quelque moyen que ce soit, mécanique, électronique, photocopie, enregistrement ou autre, sans l'autorisation écrite préalable de l'éditeur.
Pour information, contactez-nous au:
Veeda Recherche Clinique Privée Limitée
Complexe Vedant, à côté du club YMCA, SG Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat Inde.
Téléphone: + 91-79-3001-3000
Fax: + 91-79-3001-3010
Courriel : info@veedacr.com


{kind=link}