Ilościowe biomarkery farmakodynamiczne: wpływ leków i bioanaliza w rozwoju nowych jednostek chemicznych
11 stycznia 2024 r.
Strona główna > Spostrzeżenia Veedy > Integralność danych w badaniach klinicznych

Społeczność naukowa była świadkiem kilku z najgorszych tragedii w historii integralności danych z badań klinicznych. Od 2015 roku do chwili obecnej czasopisma The Journal of the American Medical Association (JAMA) i JAMA Network opublikowały co najmniej 18 zawiadomień zawierających obawy związane z błędami i/lub fałszowaniem danych.
1 Na przykład badania przeprowadzone przez japońskiego anestezjologa i badacza w leczeniu nudności i wymiotów pooperacyjnych zostały poddane przeglądowi przez Japońskie Towarzystwo Anestezjologów (JSA) w 2012 roku i przyniosły zaskakujące odkrycia.
Dane uzyskane z badań były albo całkowicie sfabrykowane, albo sfałszowane, a około 210 prac opublikowanych przez anestezjologów zawierało sfałszowane dane.
2 Luki w integralności danych spowodowały znaczną utratę przychodów, a koszty bezpośrednie oszacowano na blisko 525,000 1.3 dolarów amerykańskich, a koszty pośrednie na około XNUMX miliona dolarów amerykańskich.3
Takie niewłaściwe postępowanie naukowe pobudziło do zaostrzenia przepisów i przepisów mających na celu monitorowanie opracowywania i zażywania narkotyków.
Naukowcy uznali potrzebę integralności danych na każdym etapie, aby chronić ludzi, począwszy od rozwój przedkliniczny do nadzoru nad bezpieczeństwem farmakoterapii.
Integralność danych definiuje się jako dane w formie papierowej lub elektronicznej, które są kompletne, dokładne, spójne i niezawodne w całym cyklu życia, od momentu utworzenia danych, archiwizacji, skanowania, przechowywania i zniszczenia.
4 Zaktualizowane wytyczne Międzynarodowej Rady ds. Harmonizacji Dobrej Praktyki Klinicznej (ICH GCP E6[R2]) ponownie podkreślają potrzebę integralności danych, a także znaczenie monitorowania danych klinicznych przez cały czas trwania badania.
Amerykańska Agencja ds. Żywności i Leków (FDA) używa akronimu ALCOA do określenia oczekiwań w zakresie integralności danych.4
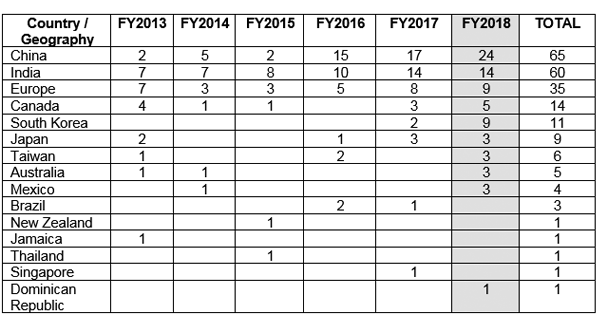
FDA wydała listy ostrzegawcze dotyczące dobrych praktyk produkcyjnych (GMP) do różnych krajów poza Stanami Zjednoczonymi (USA), powołując się na problemy związane ze zgodnością z integralnością danych. Rysunek 1 pokazuje, że Chiny otrzymały maksymalną liczbę pism ostrzegawczych GMP, a za nimi plasują się Indie i Europa.5
Rysunek 1. Listy ostrzegawcze GMP wydawane poza Stanami Zjednoczonymi5
Integralność danych można monitorować poprzez kontrolę następujących obszarów:6
SDV
Ścisłe przestrzeganie dobrych praktyk dokumentowania (GDP) w dokumentacji badań klinicznych jest sposobem na zapewnienie integralności danych. PKB należy stosować w przypadku zapisów papierowych, a także zapisów elektronicznych i podpisów.
Równie ważna jest konieczność przechowywania i porządkowania niezbędnych dokumentów wymaganych przed rozpoczęciem badania klinicznego, w jego trakcie oraz po jego zakończeniu lub zakończeniu.
Zbiór niezbędnych dokumentów przechowywanych w ośrodku sponsora i ośrodku badacza nazywany jest głównym aktem badania klinicznego (TMF). TMF odgrywa główną rolę w ułatwianiu prowadzenia badań klinicznych i zarządzania nimi, umożliwiając w ten sposób integralność danych i zgodność z GCP na wszystkich etapach badania klinicznego. TMF to dokument poddawany przeglądowi podczas audytu lub inspekcji.8
Wiele firm farmaceutycznych zmierza obecnie w stronę elektronicznego TMF (e-TMF), aby ułatwić zarządzanie dużymi i złożonymi badaniami klinicznymi, w które angażuje się wiele działów lub CRO.
Dostęp do danych i kontrola
Należy zachować ostrożność podczas obchodzenia się z danymi z badań klinicznych. Poufność danych należy zachować na wszystkich etapach badania klinicznego, łącznie z wynikami danych przejściowych.
9 Możliwość manipulowania danymi, np. zmiany, usuwania lub fałszowania danych, powinna być ograniczona poprzez wyraźne rozgraniczenie ról. Zapobiega to również potencjalnym konfliktom interesów między podobnymi rolami, które mogą utrudniać integralność danych.4
Narodowy Instytut Zdrowia (NIH) stwierdza, że jedynie członkowie Rady ds. Monitorowania Danych i Bezpieczeństwa (DSMB) z prawem głosu powinni mieć możliwość przeglądania wyników analiz okresowych, chyba że okoliczności wymagają udostępnienia danych, na przykład w przypadku poważnych niekorzystnych skutków wydarzenia.
9Ponadto członkowie DMC nie powinni mieć żadnego konfliktu interesów, który miałby wpływ na dane końcowe. FDA zaleciła również stosowanie modelu „niezależnego statystyka” do analizy danych tymczasowych, który jest niezależny od głównego badacza i sponsora badania i zgłasza bezstronne wyniki DMC.10
Monitorowanie danych
Konieczne jest powołanie niezależnego komitetu monitorującego dane (DMC), dla którego priorytetem będzie bezpieczeństwo i interesy włączonych uczestników oraz sprawdza autentyczność danych, a także przebieg badania klinicznego.9
Monitorowanie na miejscu: ma na celu wyśledzenie ewentualnych rozbieżności pomiędzy danymi źródłowymi a danymi wprowadzonymi. Szczególnie przydatne jest również sprawdzenie, czy personel ośrodka zaznajomił się z dokumentem badawczym i czy wykazał się odpowiedzialnością za przeprowadzenie badania w sposób etyczny i odpowiedzialny.11
Scentralizowane podejście oparte na ryzyku: ICH GCP E6(R2) podkreśla potrzebę scentralizowanego monitorowania w celu zmniejszenia liczby wizyt kontrolnych monitora klinicznego i umożliwienia zdalnej lokalizacji wiarygodnych i niewiarygodnych danych przez statystyków lub inny personel zarządzający danymi.4,11
Monitorowanie oparte na ryzyku: Firma sponsorująca ma obowiązek opracować solidny plan zarządzania ryzykiem, aby zapobiegać ryzyku dla ludzi lub je ograniczać poprzez nadzorowanie przebiegu badania i monitorowanie jakości danych we wszystkich ośrodkach badawczych.11
Audyty integralności danych12
Aby uniknąć ogromnych konsekwencji finansowych i strat w działalności, firmy sponsorujące i CRO powinny położyć wystarczający nacisk na utrzymanie integralności danych na każdym etapie badania klinicznego, aby zapewnić ich kompletność, dokładność i spójność.
Źródła
1. Bauchner H., Fontanarosa Phil B., Flanagin A i wsp. Niewłaściwe postępowanie naukowe i czasopisma medyczne. 2018;320(19):1985-1987 https://jamanetwork.com/journals/jama/fullarticle/2708590
2. George SL i Buyse M. Oszustwa związane z danymi w badaniach klinicznych. Clin Investig (Londyn). 2015; 5 (2): 161 – 173.
3. Michalek AM, Hutson AD, Wicher CP i wsp. Koszty i niedoceniane konsekwencje niewłaściwego postępowania badawczego: studium przypadku. PLoS Med. 2018;7(8):e1000318. https://doi.org/10.1371/journal.pmed.1000318
4. Rutherford M. ICH E6(R2) i integralność danych: cztery kluczowe zasady. Badacz kliniczny. 2018 April;32(4):doi:10.14524/CR-18-4021. https://acrpnet.org/2018/04/17/ich-e6r2-data-integrity-four-key-principles/
5. https://www.pharmaceuticalonline.com/doc/an-analysis-of-fda-fy-drug-gmp-warning-letters-0003 Dostęp 26 kwietnia 2019 r
6. Moody LE i McMillan S. Utrzymanie integralności danych w randomizowanych badaniach klinicznych. Nur Res. 2002 Mar-Apr;51(2):129-33. https://www.ncbi.nlm.nih.gov/pubmed/11984384
7. http://firstclinical.com/fda-gcp/?show=MonitoringvAuditing&search=compliance&type=&page=1 Dostęp 26 kwietnia 2019 r
8. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-good-clinical-practice-compliance-relation-trial-master-file-paper/electronic-content-management-archiving-audit-inspection-clinical-trials_en.pdf Dostęp 26 kwietnia 2019 r
9. Fleming TR, Sharples K, McCall J i wsp. Utrzymanie poufności danych tymczasowych w celu zwiększenia integralności i wiarygodności badania. Próby Clin. 2008;5(2):157-67. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2703711/
10. Ellenberg SS. Ochrona uczestników badań klinicznych i ochrona integralności danych: czy sprostamy wyzwaniom? PLoS Med. Czerwiec 2012; 9(6):e1001234.
11. https://www.thefdagroup.com/thefdgroup-blog/conducting-data-integrity-audits-a-quick-guide Dostęp 26 kwietnia 2019 r.
Informacje zawarte w tym artykule mają na celu wyłącznie zapewnienie ogólnych wskazówek w sprawach będących przedmiotem zainteresowania do osobistego użytku czytelnika, który przyjmuje pełną odpowiedzialność za ich wykorzystanie. W związku z tym informacje dotyczące tego artykułu są podawane przy założeniu, że autor(zy) i wydawca(zy) nie są w tym dokumencie zaangażowani w świadczenie profesjonalnych porad ani usług. W związku z tym nie należy go stosować jako substytutu konsultacji z kompetentnym doradcą. Przed podjęciem jakiejkolwiek decyzji lub podjęciem jakichkolwiek działań, czytelnik powinien zawsze skonsultować się z profesjonalnym doradcą w sprawie opublikowania odpowiedniego artykułu.
Chociaż dołożono wszelkich starań, aby informacje zawarte w tym artykule zostały uzyskane z wiarygodnych źródeł, Veeda Clinical Research nie ponosi odpowiedzialności za jakiekolwiek błędy lub pominięcia ani za wyniki uzyskane w wyniku wykorzystania tych informacji.
Wszystkie informacje zawarte w tym artykule są dostarczane w stanie „takim, jakie są”, bez gwarancji kompletności, dokładności, aktualności ani wyników uzyskanych w wyniku wykorzystania tych informacji, ani bez jakiejkolwiek gwarancji, wyraźnej lub dorozumianej, w tym między innymi do gwarancji wydajności, przydatności handlowej i przydatności do określonego celu.
Nic w niniejszym dokumencie nie zastąpi w żadnym stopniu niezależnych badań oraz rzetelnej oceny technicznej i biznesowej czytelnika. W żadnym wypadku firma Veeda Clinical Research ani jej partnerzy, pracownicy lub agenci nie ponoszą odpowiedzialności wobec czytelnika ani nikogo innego za jakiekolwiek decyzje lub działania podjęte w oparciu o informacje zawarte w tym artykule ani za jakiekolwiek szkody wtórne, szczególne lub podobne nawet jeśli został poinformowany o możliwości wystąpienia takich szkód.
Żadna część tej publikacji nie może być powielana, przechowywana w systemie wyszukiwania ani przesyłana w jakiejkolwiek formie i w jakikolwiek sposób, mechaniczny, elektroniczny, poprzez kserowanie, nagrywanie lub w inny sposób, bez uprzedniej pisemnej zgody wydawcy.
Aby uzyskać informacje, skontaktuj się z nami pod adresem:
Veeda Badania kliniczne prywatna spółka z ograniczoną odpowiedzialnością
Kompleks Vedant, obok klubu YMCA, autostrada SG,
Vejalpur, Ahmadabad – 380 051,
Indie Gujarat.
Telefon: + 91-79-3001-3000
Faks: + 91-79-3001-3010
E-mail: info@veedacr.com

11 stycznia 2024 r.

27 lipca 2023 r.

21 czerwca 2023 r.

8 stycznia 2022 r.

1 grudnia 2021 r.
