
Indie wyłaniają się jako kraj o ogromnym potencjale wniesienia wkładu w krajowe i międzynarodowe platformy badań klinicznych.
Połączenia Centralna Organizacja Kontroli Standardów Narkotyków (CDSCO) jest krajowym organem regulacyjnym Indii, zarządzanym przez Generalnego Inspektora Kontroli Leków w Indiach (DCGI).1,2,3
DCGI jest odpowiedzialna za koordynację inspekcji sponsorów, jednostek produkcyjnych, a także miejsc szlaków.3
Wczesne lata rozwoju klinicznego
W 2000 r. Indyjska Rada ds. Badań Medycznych (ICMR) ustaliła wytyczne etyczne dotyczące prowadzenia badań biomedycznych na ludziach.4 W roku 2005 dokonano zmiany załącznika Y do ustawy o lekach i kosmetykach z 1945 r., aby dostosować indyjskie przepisy regulacyjne do definicji i procedur akceptowanych na szczeblu międzynarodowym.
Zmiany obejmowały:
- Definiowanie faza I do IV fazy badania
- Wyznaczone obowiązki sponsora(ów) i badacza(ów)
- Opcje rejestrowania wszelkich odchyleń lub zmian w zatwierdzonym protokole badania
Indie podpisały także w 2005 r. umowę dotyczącą handlowych praw własności intelektualnej (TRIPS), aby otworzyć perspektywy prowadzenia większej liczby badań klinicznych w Indiach.5
Oprócz harmonizacji aktów prawnych do standardów międzynarodowych, Indie szybko stały się korzystnym kierunkiem badań klinicznych, ponieważ oferowały:6
- Anglojęzyczni specjaliści w służbie zdrowia
- Ekspertyza techniczna
- Rosnąca ekonomia
- Technologia światowej klasy
- Duża, zróżnicowana i nieleczona populacja
Odstaw na badania kliniczne
Pomimo zmian w przepisach wiele międzynarodowych firm farmaceutycznych wykorzystało dużą populację, która albo miała niewystarczającą wiedzę na temat badań klinicznych, albo była analfabetą. Ponadto źle zdefiniowany system opieki zdrowotnej zwiększył wyzwania związane z monitorowaniem nieetycznych praktyk.
Doprowadziło to do prowadzenia badań klinicznych pod niewielkim nadzorem i do braku rejestrowania świadomej zgody pacjenta w formie pisemnej lub audiowizualnej.
Pacjentom podawano badane leki lub wyroby bez ujawnienia znanych poważnych działań niepożądanych, niektóre prowadzących do śmierci uczestników. Co więcej, nie powołano niezależnej komisji śledczej, która miałaby ustalić, czy śmierć pacjenta była związana/niezwiązana z badanym produktem lub wyrobem.4
Lata 2010–2013 to okres próby w branży Indyjskie badanie kliniczne scenariusza ze względu na skumulowane złe skutki prowadzenia nieetycznych procesów.
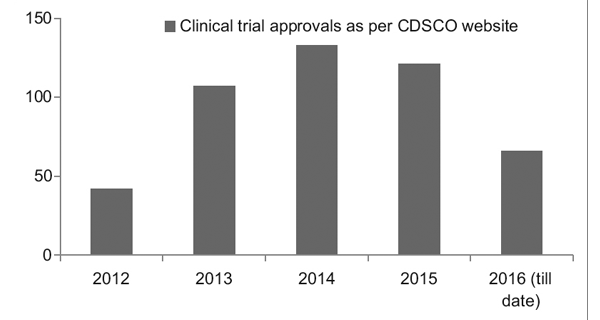
Jednakże dzięki lepszym ramom regulacyjnym indyjski rejestr badań klinicznych (CTRI) odnotowuje stały wzrost liczby prowadzonych badań, jak widać na ryc. 1. Zaobserwowano również, że większość badań to badania III fazy próby.7
Postać 1: Trendy w badaniach klinicznych na przestrzeni lat
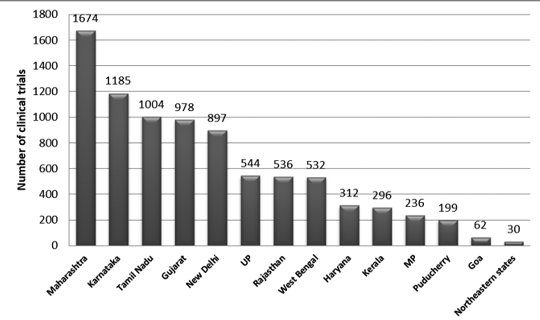
Rysunek 2 przedstawia rozkład prób w Indiach w latach 2007–2015 według stanów. W tym okresie zarejestrowano około 3330 szlaków.
Zaobserwowano, że najwięcej prób przeprowadzono w Maharasztrze, a najmniej w stanie północno-wschodnim. Wśród stanów północno-wschodnich w Nagaland nie przeprowadzono żadnych badań.7
Postać 2: Rozkład badań klinicznych na szczeblu stanowym w Indiach (dane z lat 2007–2015)7
Ożywienie scenariusza klinicznego i regulacyjnego
W 2014 r. CDSCO utworzyło 12 nowych komitetów doradczych ds. narkotyków (NDAC) i 25 komitetów ekspertów przedmiotowych (SEC). W skład tych komitetów wchodzi wielu ekspertów z wybitnych uczelni i instytucji rządowych, których zadaniem jest skrócenie terminu zatwierdzania badania klinicznego do 6–7 miesięcy.
Proces trójstopniowy składa się z:9
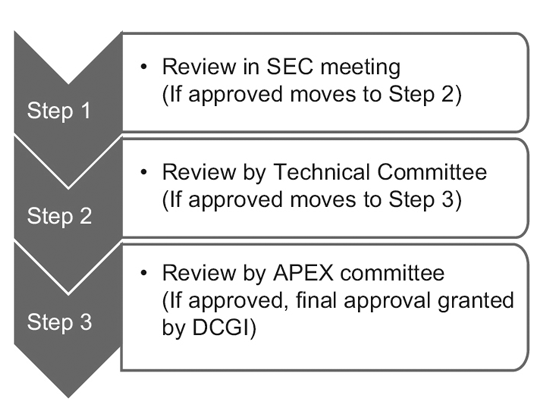
Jednakże wnioski o globalne badania kliniczne rozpatruje wyłącznie SEC i nie jest wymagana żadna dalsza zgoda ze strony Komitetu Technicznego ani Komitetu Apex. Wnioski badawcze dotyczące nowych leków (IND) są również rozpatrywane niezależnie przez komisję IND i nie wymagają zatwierdzenia przez komisję Apex.
Komisja techniczna wchodzi w grę tylko wtedy, gdy SEC odrzuciła wniosek sponsora, a sponsor czuje się pokrzywdzony decyzją. W takim przypadku, jeśli Komitet Techniczny nie zgodzi się z decyzją SEC, ma prawo uchylić decyzję SEC.10
W marcu 2019 r. Ministerstwo Zdrowia i Opieki Rodzinnej Indii wydało rozporządzenie Nowe leki i zasady badań klinicznych 2019 z zamiarem przyspieszenia zatwierdzania badań klinicznych, nowych leków, badań biorównoważności (BE) lub biodostępności (BA).
W zasadach tych usunięto również wszelkie niejasności istniejące w odniesieniu do przepisów Komisji ds. Etyki (KE).11
Najważniejsze informacje dotyczące nowych leków i zasad badań klinicznych, 201911 |
Zaktualizowany regulamin11 |
Harmonogram zatwierdzania badań klinicznych |
90 dni roboczych od otrzymania wniosku w przypadku leków wykrytych poza Indiami i 30 dni roboczych w przypadku nowych leków lub IND w Indiach |
Produkcja nowych leków lub badania IND, BE i BA lub analiza testów lub badanie |
Wymagane jest pozwolenie od Central Licensing Authority (CLA) |
Odstąpienie od lokalnych badań klinicznych |
· Jeśli CLA zatwierdziło wprowadzenie do obrotu nowego leku w innych krajach lub udzieliło pozwolenia na prowadzenie globalnych badań klinicznych nowego leku w Indiach · Brak dowodów na różnicę w metabolizmie, bezpieczeństwie lub skuteczności ze względu na różnicę w profilu genetycznym populacji Indii |
Okres ważności badania klinicznego |
2 lata od daty wydania przez CLA |
Dostęp po okresie próbnym do IND lub nowego leku |
W wyjątkowych okolicznościach lek ma być bezpłatnie dystrybuowany wśród uczestników badania zgodnie z zaleceniami CLA, ale sponsor nie ponosi żadnej odpowiedzialności za użycie leku po zakończeniu badania. |
Spotkania przed i po złożeniu wniosku |
Aby uzyskać wskazówki dotyczące prawa i procedur regulujących proces produkcji i licencjonowania lub udzielania zezwoleń. |
Dopuszczenie do badań prowadzonych przez KE i rejestracja KE |
· Należy uzyskać zgodę KE innego ośrodka badawczego, jeśli ośrodek badawczy nie posiada EC, a EC powinien znajdować się w promieniu 50 km od miejsca badawczego.· Rejestracja WE zatwierdzona przez CLA pozostaje ważna przez pięć lat od daty wydania. |
Warunki, jakie należy spełnić, aby przeprowadzić badanie kliniczne |
· Składanie raportu o stanie co kwartał lub w zależności od czasu trwania badania w celu śledzenia rejestracji uczestników· Raportowanie online statusu badania klinicznego co sześć miesięcy za pośrednictwem portalu SUGHAM, aby wiedzieć, czy badanie jest w toku, czy zostało zakończone, czy też zostało zakończone. |
Opłata za nabycie licencji, zaświadczenia o rejestracji i zezwolenia na okres próbny |
Różne struktury opłat w zależności od celu okresu próbnego. Opłata od 50,000 5,00,000 do XNUMX XNUMX XNUMX INR. |
Pokonywanie barier
Wyzwania związane z badaniami klinicznymi są wieloaspektowe i wiążą się z przestrzeganiem ram regulacyjnych w odpowiedzialny i etyczny sposób przez zainteresowane strony, rząd i system sądowy.
Bezpieczeństwo i ochrona pacjenta sprawą najwyższej wagi powinno być ustanowienie ścisłych zasad dotyczących:12
- świadomej zgody w drodze nagrania audiowizualnego i w języku, który jest dla pacjenta komfortowy
- szacunek dla pochodzenia kulturowego, społecznego, ekonomicznego i edukacyjnego pacjenta
- terminowe zgłaszanie SAE
Nowe podstawowe zasady które mogą otworzyć możliwość rozszerzenia badań medycznych w Indiach to:13
- zatwierdzenie propozycji złożonych do DCGI w ciągu 30 dni od złożenia wniosku, jeżeli DCGI nie otrzyma żadnej wiadomości
- szybkie śledzenie krajowych zezwoleń
- spotkania z komitetem ekspertów przed i po złożeniu wniosku w celu zapewnienia większej przejrzystości procesu i ustalenia dobrze określonego harmonogramu zakończenia badania
- odszkodowanie za próbę w przypadku, gdy badany lek doprowadził do SAE/śmierci.
Wykwalifikowana kadra i najnowocześniejsza infrastruktura odgrywają również ważną rolę w przyciąganiu firm sponsorujących. Badania wykazały, że chociaż badania fazy III są prowadzone na szeroką skalę w Indiach, badania fazy I wydają się ograniczać do kraju sponsora.
Można to przypisać obawom Sponsora przed pozyskaniem wykwalifikowanej siły roboczej i technologii. Aby umożliwić prowadzenie rodzimych badań w Indiach, kluczowe znaczenie ma zapewnienie personelowi odpowiedniego narażenia lub ustawicznego kształcenia medycznego oraz dostęp do najnowocześniejszych technologii, aby zostać uznanym za kraj wystarczająco kompetentny do przeprowadzenia jakichkolwiek prób fazowych.9
Równie ważna jest potrzeba dostępności wykwalifikowanych pracowników służby zdrowia w całym kraju, aby uwzględnić nierówny rozkład badań klinicznych w poszczególnych stanach.
Koncentrowanie badania na konkretnym stanie może prowadzić do stronniczych wniosków i nadmiernego upraszczania lub wyolbrzymiania obciążenia lub stanu chorobowego. Zapewniając dostęp do badania klinicznego osobom ze wszystkich stanów, nie tylko minimalizujemy stronniczość, ale także uwzględniamy zróżnicowane populacje etniczne.9
Przyszłość
Dzięki pozytywnym, przyjaznym pacjentom, szybkim i przejrzystym przepisom regulacyjnym Indie będą w dalszym ciągu rozwijać się jako międzynarodowe centrum testowania i opracowywania innowacyjnych leków i wyrobów medycznych.
Źródła
1. Evangeline L, Mounica NVN, Reddy VS i wsp. Proces regulacyjny i etyka badań klinicznych w Indiach (CDSCO). Dziennik innowacji farmaceutycznych. 2017;6(4):165-9. http://www.thepharmajournal.com/archives/2017/vol6issue4/PartC/6-4-4-176.pdf
2. Lahiry S., Sinha R., Choudhary S i wsp. Zmiana paradygmatu w przepisach dotyczących badań klinicznych w Indiach. Indyjski Dziennik Reumatologii, 2018; 13: 51-5.
3. Gogtay NJ, Ravi R i Thatte UM. Wymogi regulacyjne dotyczące badań klinicznych w Indiach: co powinni wiedzieć naukowcy. Indian Journal of Anesthesia. 2017 Mar;61(3):192-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5372399/
4. Ramu B, Kumar MS i Ramakrishna N. Aktualny scenariusz regulacyjny dotyczący prowadzenia badań klinicznych w Indiach. Sprawy regulacyjne branży farmaceutycznej. Otwarty dostęp, 2015; 4: 2. https://www.researchgate.net/publication/281765214_Current_Regulatory_Scenario_for_Conducting_Clinical_Trials_in_India
5. Burt T., Sharma P., Dhillon S i wsp. Środowisko badań klinicznych w Indiach: wyzwania i proponowane rozwiązania. Journal of Clinical Research Bioethics. 2014;5:6. DOI: 10.4172/2155-9627.1000201
6. Chaturvedi M, Gogtay NJ, Thatte UM. Czy badania kliniczne prowadzone w Indiach odpowiadają ich potrzebom w zakresie opieki zdrowotnej? Audyt Rejestru Badań Klinicznych Indii. Perspektywy w badaniach klinicznych. 2017;8(4):172-5.
7. http://ctri.nic.in/Clinicaltrials/news/CTRI_Newsbulletin_July-Dec_2017.pdf Dostęp: 23 kwietnia 2019 r.
8. Bhave A i Menon S. Otoczenie regulacyjne badań klinicznych: niedawna przeszłość i oczekiwana przyszłość. Perspektywy w badaniach klinicznych, 2017; 8: 11.6.
9. Key Highlights of New Drugs & Clinical Trial Rules, 2019. Dostęp: 23 kwietnia 2019 r.
10. Dan S., Karmakar S., Ghosh B i wsp. Cyfryzacja badań klinicznych w Indiach: nowy krok CDSCO w kierunku zapewnienia wiarygodności danych i bezpieczeństwa pacjentów. Farmaceutyczne kwestie regulacyjne: otwarty dostęp. 2015;4(3): DOI: 10.4172/2167-7689.1000149.
11. https://www.thehindubusinessline.com/news/new-rules-sweeten-the-deal-for-clinical-trials-by-indian-pharma-cos/article26283499.ece Dostęp 23 kwietnia 2019 r.
Zrzeczenie się:
Informacje zawarte w tym artykule mają na celu wyłącznie dostarczenie ogólnych wskazówek w sprawach będących przedmiotem zainteresowania do osobistego użytku czytelnika, który przyjmuje pełną odpowiedzialność za ich wykorzystanie. W związku z tym informacje zawarte w tym artykule są podawane przy założeniu, że autor(zy) i wydawca(zy) nie są tutaj zaangażowani w świadczenie profesjonalnych porad ani usług.
W związku z tym nie należy go stosować jako substytutu konsultacji z kompetentnym doradcą. Przed podjęciem jakiejkolwiek decyzji lub podjęciem jakichkolwiek działań, czytelnik powinien zawsze skonsultować się z profesjonalnym doradcą w sprawie opublikowania odpowiedniego artykułu.
Chociaż dołożono wszelkich starań, aby informacje zawarte w tym artykule zostały uzyskane z wiarygodnych źródeł, Veeda Clinical Research nie ponosi odpowiedzialności za jakiekolwiek błędy lub pominięcia ani za wyniki uzyskane w wyniku wykorzystania tych informacji.
Wszystkie informacje zawarte w tym artykule są dostarczane w stanie „takim, jakie są”, bez gwarancji kompletności, dokładności, aktualności ani wyników uzyskanych w wyniku wykorzystania tych informacji, ani bez jakiejkolwiek gwarancji, wyraźnej lub dorozumianej, w tym między innymi do gwarancji wydajności, przydatności handlowej i przydatności do określonego celu.
Nic w niniejszym dokumencie nie zastąpi w żadnym stopniu niezależnych badań oraz rzetelnej oceny technicznej i biznesowej czytelnika. W żadnym wypadku nie będzie Badania kliniczne Veedalub jej partnerzy, pracownicy lub agenci nie ponoszą odpowiedzialności wobec czytelnika lub kogokolwiek innego za jakiekolwiek decyzje podjęte w oparciu o informacje zawarte w tym artykule lub za jakiekolwiek szkody wtórne, szczególne lub podobne, nawet jeśli zostali powiadomieni o możliwości takich szkód.
Żadna część tej publikacji nie może być powielana, przechowywana w systemie wyszukiwania ani przesyłana w jakiejkolwiek formie i w jakikolwiek sposób, mechaniczny, elektroniczny, poprzez kserowanie, nagrywanie lub w inny sposób, bez uprzedniej pisemnej zgody wydawcy.
Aby uzyskać informacje, skontaktuj się z nami pod adresem:
Veeda Badania kliniczne prywatna spółka z ograniczoną odpowiedzialnością
Kompleks Vedant, obok klubu YMCA, autostrada SG,
Vejalpur, Ahmadabad – 380 051,
Indie Gujarat.
Telefon: + 91-79-3001-3000
Faks: + 91-79-3001-3010
E-mail: info@veedacr.com


{kind=link}