생물학적 분석은 표적 식별부터 납 화합물 발견까지 약물 발견의 각 단계에 관여합니다. 생물검정은 조사 중인 약물의 치료 효능을 보여주는 귀중한 정보를 제공합니다.
생물학적 검정 중에 생성된 데이터는 의약품 개발 및 완제품 생물학적 제품의 품질 관리에도 중요한 역할을 합니다. 적절하게 설계된 생물학적 검정은 적합한 생물학적 시스템에 대한 참조 또는 표준과 비교할 때 생물학적 표적(단백질)에 대한 의약품 또는 생물학적 제제의 생물학적 효과, 활성, 신호 전달 과정 및 수용체 결합 능력을 평가하는 데 도움이 됩니다.
신약 발견 및 개발에 참여하는 제약 및 생명공학 회사는 다양한 잠재적 메커니즘 분석을 위해 생물학적으로 관련된 분석법을 개발하는 데 지속적으로 어려움을 겪고 있습니다.
이 과정에는 품질이 중요한 시약의 사용, 특정 세포주의 사용, 정제된 테스트 약물 및 참조 의약품이 포함되며, 이는 때때로 제약이 될 수 있습니다. 이러한 활동의 대부분은 충분한 시간이 필요하며 이는 바이오제약 제조업체에게 제한 요소가 될 수 있습니다.
아웃소싱 활동을 수행할 가치가 있습니다. 평판이 좋은 CRO 서비스 제공업체 개발 노력에 소요되는 시간을 절약하고 의약품의 기능적 활성에 대해 편견 없는 의견을 갖기 위함입니다.
Veeda Group은 기업을 위한 생물검정을 설계, 개발, 실행 및 검증할 수 있는 자격을 갖추고 경험이 풍부한 과학자를 보유하고 있으며 최고의 생물검정 서비스를 제공합니다(체외에서 와 생체내에서)는 제약 및 생명공학 회사의 신약 발견 및 개발 여정을 지원하기 위해 의미 있는 데이터를 생성합니다.
Veeda Group의 생물검정 개발 및 실행 경험은 다음과 같습니다.
- 플라크 감소 중화 시험(PRNT 분석)
- 체외에서 피부 감작성 인간 세포주 활성화 시험(h-CLAT 분석)
- Nab 분석
- 분석법 개발(약력학, 약동학, 면역원성 및 바이오마커 평가)
- 생체 내 황체형성호르몬, 에포에틴, HCG, 재조합 FSH, β-HCG 및 인슐린과 같은 약물 분자에 대한 생물학적 검정.
- 바이오시밀러에 대한 ADCC 분석 및 기타 다른 분석 엑스 비보 분석, 세포 기반 분석, 수용체 결합 분석, 사이토카인 방출 분석 및 ADA 분석.
Veeda Group은 다양한 기술 플랫폼을 통해 통합 검색, 개발 및 규제 서비스를 제공합니다.
- 탐색적 독성학 연구
- 규제 독성학 연구
- 체외 생물학적 검정
- 생체 외 생물학적 검정
이 그룹은 또한 치료용 단일클론 항체, 인슐린 및 인슐린 유사체, 사이토카인, 저분자량 헤파린, 바이오시 밀러, 호르몬 및 바이오마커.
Veeda 그룹은 박테리아 또는 포유류 숙주 발현 시스템에서 유래된 비당화 단백질 및 당단백질과 같은 재조합 단백질을 개발할 수 있는 능력을 입증했습니다.
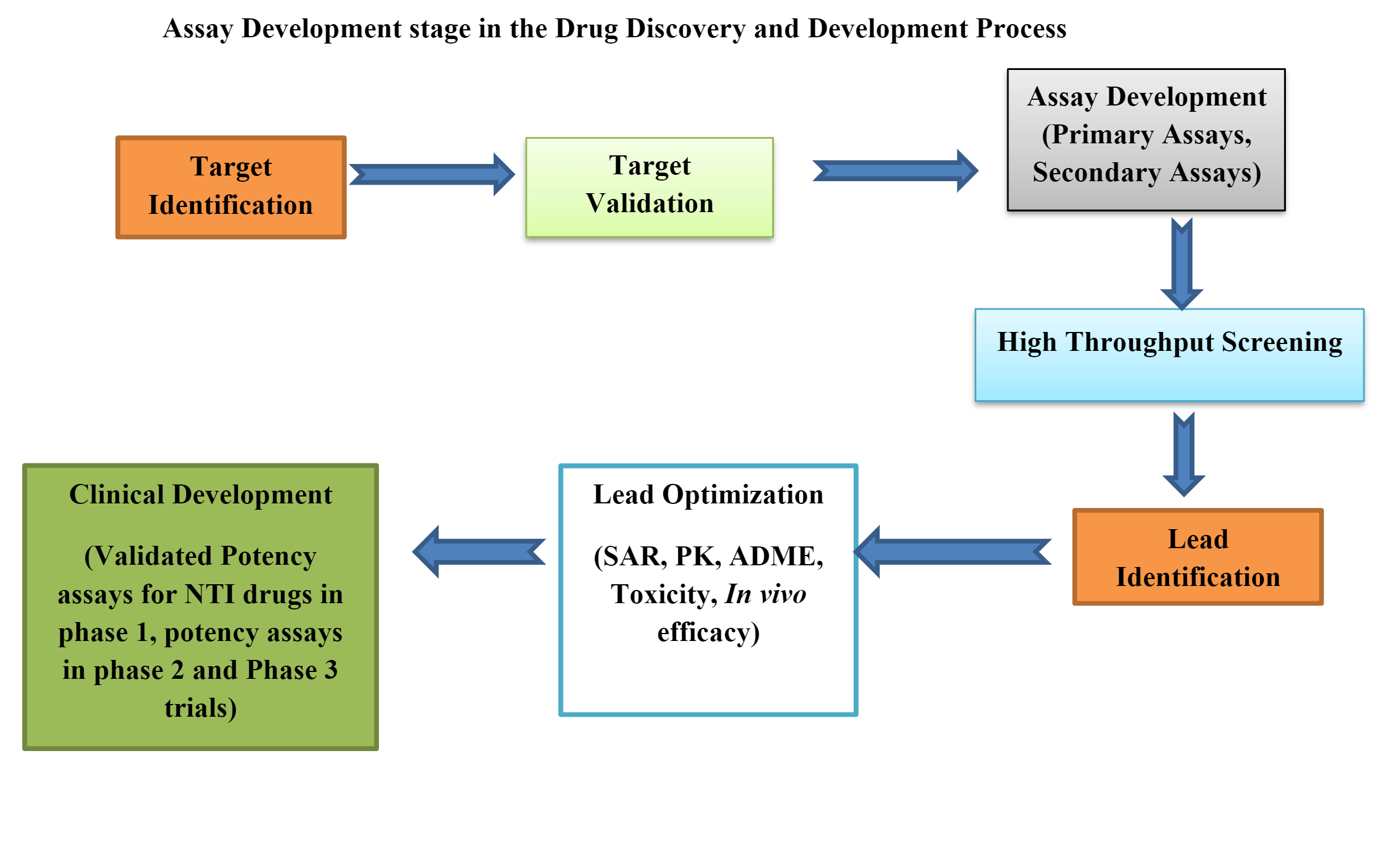
전임상 약물 개발에서의 생물학적 검정
생물학적 분석 또는 생물학적 분석은 다음과 같은 경우에 필수적인 도구입니다. 전임상 약물 개발. 전임상 생물학적 분석은 다음과 같습니다. 생체 내, 생체 외및 체외에서.
생체 내 생물검정은 통계 도구, 연구별 실험실 기법, 잘 설계된 연구 프로토콜 준수와 함께 참조 의약품 또는 정의된 효능의 표준 물질을 사용한 테스트의 기능적 효과에 대한 보다 현실적이고 예측 가능한 측정을 제공합니다.
이러한 분석은 기존 분석보다 더 나은 표적 참여, 대사 및 신약의 약동학의 복잡성을 포착합니다. 체외에서 생물검정.
i에서 가장 일반적으로 사용되는 실험 포유류엔비보 효능 분석은 생쥐와 쥐입니다. 때로는 분석의 민감도 및 적합성에 따라 다른 종을 사용할 수도 있습니다.
생물검정의 개발 및 검증
생물검정은 일련의 화합물에서 원하는 생물학적 활성을 나타내는 신호를 식별하기 위한 스크리닝 방법으로 사용됩니다. 일반적으로 생물검정을 통해 두 가지 다른 유형의 신호, 즉 선형 용량-반응과 S자형(S자형) 용량-반응이 생성될 수 있습니다.
하나의 솔루션이 모든 생물학적 분석에 적합하지 않기 때문에 데이터를 평가하고 분석하여 각 생물학적 분석을 수행하기 위한 정확한 접근 방식을 개발하는 것이 좋습니다.
생물검정의 수명주기 단계는 다음과 같이 구분됩니다.
1단계: 분석법 설계, 개발 및 최적화
2단계: 절차 수행 자격 인증
3단계: 절차 성능 검증(목적에 부합하다)
규제 요건을 충족하고 의약품을 등록하는 생물학적 분석법을 개발하는 것은 매우 복잡한 과정입니다.
생물검정 개발에는 올바른 방법을 선택하는 것과 같은 많은 전략과 전술적 설계가 포함됩니다. 생체내에서 플랫폼, 적절한 방법 또는 플레이트 설계, 데이터 분석, 시스템/샘플 지속 가능성 전략, 방법 구현, 방법 성능 및 모니터링.
용량 반응 및 곡선 맞춤 선택, 참조 개발, 효능 계산, 생물학적 분석 특성화, 생물학적 분석 계산기 설계, 생물학적 분석의 표준화 및 자동화와 같은 생물학적 분석의 개발 및 검증을 위해 따라야 할 몇 가지 단계가 있습니다. , 평가.
방법 개발과 생물학적 검정의 검증에는 세 가지 기본 영역이 포함됩니다.
- 사전 연구(식별 및 설계 단계) 검증
- 연구 중(개발 및 생산 단계) 검증
- 교차 검증 또는 방법 이전 검증
방법 개발 과정에서 잠재적인 무효 원인의 영향을 최소화하는 분석 조건과 절차가 선택됩니다. 통계적 검증에 들어갑니다. 생체내에서 분석에는 네 가지 주요 구성 요소가 포함됩니다.
- 적절한 연구 설계 및 데이터 분석 방법
- 동물의 적절한 무작위 배정
- 적절한 통계력 및 표본 크기
- 분석 실행 전반에 걸쳐 적절한 재현성.
병렬 그룹 설계, 무작위 블록 설계, 반복 측정 설계 및 교차 설계는 실험 설계의 기본 유형입니다. 생체내에서 시험.
다음은 디자인 시 염두에 두어야 할 주요 요소입니다. 생체내에서 시험:
- 모든 의미 있는 생물학적 효과(약리학적)는 통계적으로 유의해야 합니다.
- 생물학적으로 관련된 분석이 존재하지 않는 경우, 그럴듯한 효과의 범위를 고려할 수 있습니다.
- 주요 평가변수는 분석을 시작하기 전에 잘 정의되어야 합니다.
- 동물은 적절한 방식으로 치료군에 무작위로 할당되어야 합니다.
- 복용량 수준을 적절하게 선택해야 합니다. 용량 및 곡선 맞춤 선택은 생물학적 검정 개발의 가장 중요한 측면 중 하나입니다. 데이터를 맞추기 위해 신호에 사용된 모델 유형에 따라 용량이 결정됩니다. S자형 설계의 경우 4개 또는 5개의 매개변수 물류(XNUMXPL 또는 XNUMXPL) 모델이 데이터에 적합한 반면, 선형 설계의 경우 평행선 분석(PLA) 모델이 데이터에 적합합니다.
4PL 모델의 경우 XNUMX회 용량이 권장됩니다.
- 하부 점근선의 XNUMX회 선량
- 상부 점근선의 XNUMX개 선량
- 선형 범위의 세 가지 용량
대조적으로, PLA 모델의 경우 최소 XNUMX회 투여가 권장됩니다. 용량 곡선을 그리려면 최소 XNUMX회 연속 용량이 필요합니다.
- 샘플 수집을 위한 대조군과 시점의 선택은 최적이어야 합니다.
- 설계 전략은 가변성을 최소화하고 정보를 최대화해야 합니다.
설계, 개발 및 통계적 검증을 이해합니다. 생체내에서 생물검정에 대한 자세한 내용은 다음 주소로 문의해 주세요. https://www.veedacr.com. 다음 링크를 방문하면 NIH에서 언급한 지침을 읽을 수도 있습니다.
https://www.ncbi.nlm.nih.gov/books/NBK92013/pdf/Bookshelf_NBK92013.pdf
참고자료
- A. Little, “생물검정 개발의 필수 요소”, BioPharm International 32 (11) 2019
- Padmalayam, Ph.D., 신약 발견의 분석 개발
- Zwierzyna M, Overington JP (2017) 생체 내 생물학적 분석 설명의 대규모 컬렉션 분류 및 분석. PLoS Comput Biol13(7): e1005641. https://doi.org/10.1371/journal.pcbi.1005641
- White JR, Abodeely M, Ahmed S, Debauve G, Johnson E, Meyer DM, Mozier NM, Naumer M, Pepe A, Qahwash I, Rocnik E, Smith JG, Stokes ES, Talbot JJ, Wong PY. 생물의약품 등록을 지원하기 위한 생물검정 개발 모범 사례. 생명공학. 2019년 67월;3(126):137-10.2144. 도이: 2019/btn-0031-2019. Epub 5 31379198월 XNUMX일. PMID: XNUMX.
- F Chana 및 Hursh D, 제품 수명주기를 통한 생물검정: CDER 및 CBER 검토의 관점.
- Haas J, Manro J, Shannon H, 등. 생체 내 분석 지침. 2012년 1월 2012일 [1년 2004월 XNUMX일 업데이트]. In: Markossian S, Grossman A, Brimacombe K 등, 편집자. 분석 지침 매뉴얼 [인터넷]. Bethesda(MD): Eli Lilly & Company 및 국립 번역 과학 발전 센터; XNUMX-. 책장 URL: https://www.ncbi.nlm.nih.gov/books/