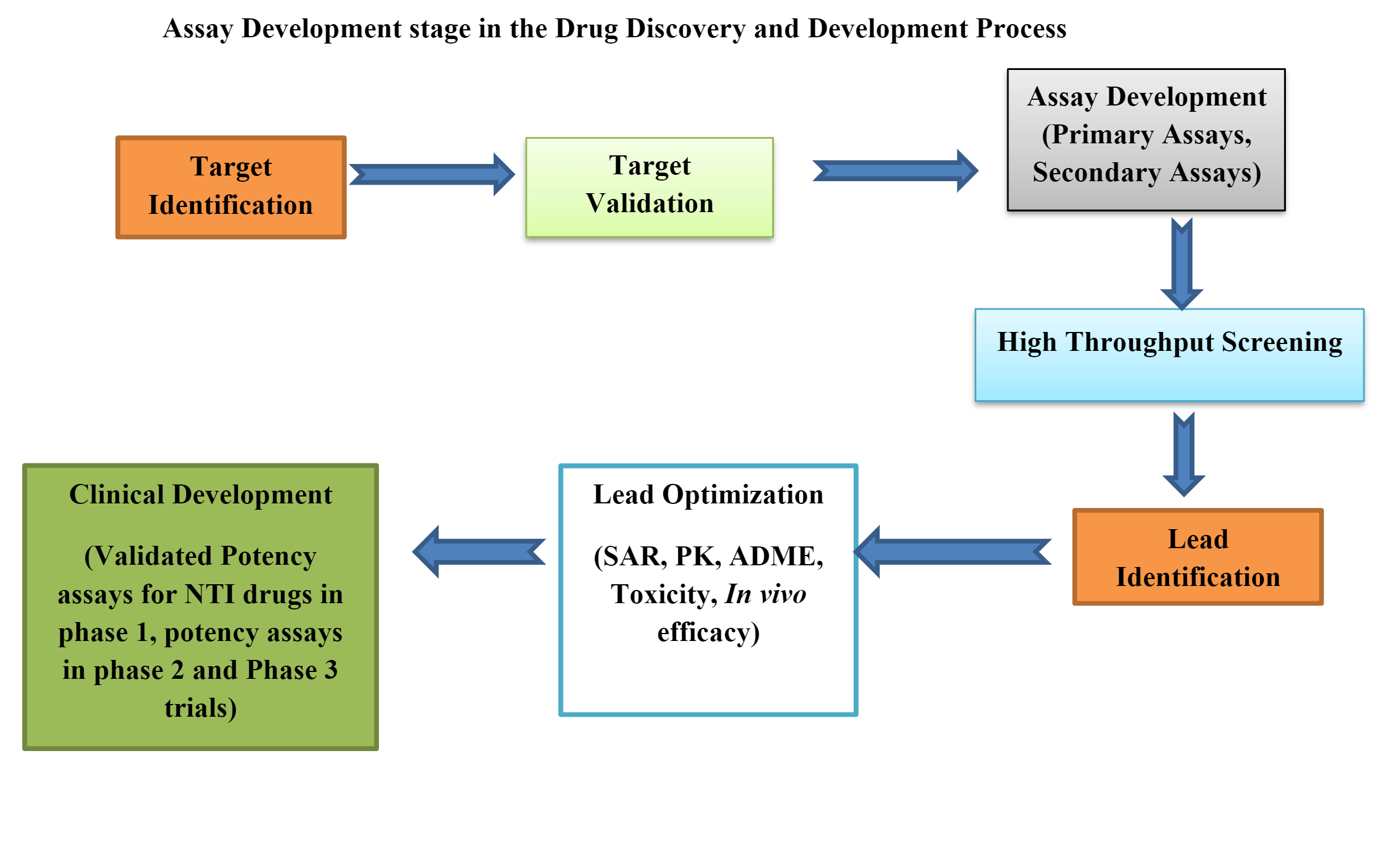
I test biologici sono coinvolti in ogni fase della scoperta di un farmaco, a partire dall'identificazione del target fino alla scoperta del composto principale. I test biologici forniscono informazioni preziose che mostrano la potenza terapeutica di un farmaco in esame.
I dati generati durante il test biologico svolgono anche un ruolo fondamentale nello sviluppo di farmaci e nel controllo di qualità dei prodotti biologici finiti. I test biologici progettati correttamente aiutano a valutare l'effetto biologico, l'attività, il processo di trasduzione del segnale e la capacità di legame del recettore del prodotto farmaceutico o biologico su un bersaglio biologico (proteine) rispetto a un riferimento o standard su un sistema biologico adatto.
Le aziende farmaceutiche e biotecnologiche coinvolte nella scoperta e nello sviluppo di farmaci sono continuamente sfidate nello sviluppo di test biologicamente rilevanti per l'analisi di molteplici potenziali meccanismi.
Il processo prevede l'uso di reagenti di qualità critica, l'uso di linee cellulari specifiche e farmaci di prova purificati e prodotti farmaceutici di riferimento che a volte possono diventare un limite. La maggior parte di queste attività richiedono tempo sufficiente, il che potrebbe diventare un fattore limitante per i produttori di prodotti biofarmaceutici.
Vale la pena esternalizzare le attività rinomati fornitori di servizi CRO per risparmiare tempo negli sforzi di sviluppo e anche per avere un'opinione imparziale sulle attività funzionali del prodotto farmaceutico.
Veeda Group dispone di scienziati qualificati ed esperti per progettare, sviluppare, eseguire e convalidare i test biologici per le aziende e fornisce servizi di test biologici di prima qualità (in vitro ed in vivo) che generano dati significativi per supportare le aziende farmaceutiche e biotecnologiche nel loro percorso di scoperta e sviluppo di farmaci.
L'esperienza di Veeda Group nello sviluppo e nell'esecuzione di test biologici include:
- Test di neutralizzazione della riduzione della placca (test PRNT)
- In Vitro Test di sensibilizzazione cutanea di attivazione della linea cellulare umana (saggio h-CLAT)
- Saggio Nab
- Sviluppo di test (farmacodinamica, farmacocinetica, immunogenicità e valutazione dei biomarcatori)
- In vivo Saggi biologici per molecole farmacologiche come l'ormone luteinizzante, l'epoetina, l'HCG, l'FSH ricombinante, la β-HCG e l'insulina.
- Test ADCC per biosimilari e diversi altri test simili Ex vivo test, test basato su cellule, test di legame del recettore, test di rilascio di citochine e test ADA.
Veeda Group fornisce servizi integrati di rilevamento, sviluppo e regolamentazione con le sue molteplici piattaforme tecnologiche:
- Studi esplorativi di tossicologia
- Studi di tossicologia normativa
- In vitro Saggi biologici
- Ex vivo Saggi biologici
Il gruppo ha anche l'esperienza per gestire una vasta gamma di prodotti bioterapici come anticorpi monoclonali terapeutici, insulina e analoghi dell'insulina, citochine, eparine a basso peso molecolare, biosimilari, Ormoni e biomarcatori.
Il gruppo Veeda ha dimostrato la capacità di sviluppare proteine ricombinanti come proteine non glicosilate e glicoproteine derivate da sistemi di espressione ospite batterici o mammiferi.
Saggi biologici nello sviluppo preclinico di farmaci
I test biologici o i test biologici sono strumenti essenziali in sviluppo di farmaci preclinici. I test biologici preclinici possono essere in vivo, ex vivoe in vitro.
in vivo i test biologici forniscono una misura più realistica e predittiva degli effetti funzionali dei test con prodotti farmaceutici di riferimento o materiale standard di potenza definita, insieme all'applicazione di strumenti statistici, tecniche di laboratorio specifiche per lo studio e aderenza al protocollo di studio ben progettato.
Questi test catturano meglio la complessità del coinvolgimento del target, del metabolismo e della farmacocinetica dei nuovi farmaci in vitro saggi biologici.
I mammiferi sperimentali più comunemente usati in iin vivo test di efficacia sono topi e ratti. Occasionalmente possono essere utilizzate altre specie a seconda della sensibilità e dell'idoneità dei test.
Sviluppo e validazione di saggi biologici
I test biologici vengono utilizzati come metodo di screening per identificare i segnali che indicano l'attività biologica desiderata da un insieme di composti. In generale, due diversi tipi di segnali possono essere generati da un saggio biologico: una risposta alla dose lineare e una risposta alla dose sigmoidale (a forma di S).
Poiché una soluzione non è adatta a tutti i test biologici, è bene valutare e analizzare i dati per sviluppare un approccio preciso per eseguire ciascun test biologico.
Le fasi del ciclo di vita di un saggio biologico si dividono in:
Fase 1: progettazione, sviluppo e ottimizzazione del metodo
Fase 2: Qualificazione delle prestazioni della procedura
Fase 3: verifica delle prestazioni della procedura (adatto allo scopo)
Lo sviluppo di un test biologico che soddisfi i requisiti normativi e ottenga la registrazione di un prodotto farmaceutico è un processo molto complesso.
Lo sviluppo di un test biologico include molte strategie e progetti tattici come la selezione del metodo corretto in vivo piattaforma, progettazione corretta del metodo o della piastra, analisi dei dati, strategia di sostenibilità del sistema/campione, implementazione del metodo, prestazioni del metodo e monitoraggio.
Ci sono diversi passaggi da seguire per lo sviluppo e la validazione dei test biologici, come la selezione della dose-risposta e dell'adattamento della curva, lo sviluppo del riferimento, il calcolo della potenza, la caratterizzazione del test biologico, la progettazione del calcolatore del test biologico, la standardizzazione e l'automazione del test biologico e infine , valutazione.
Sia lo sviluppo del metodo che la validazione dei test biologici comprendono tre aree fondamentali:
- Convalida pre-studio (fase di identificazione e progettazione).
- Convalida in studio (fase di sviluppo e produzione).
- Convalida incrociata o convalida del trasferimento del metodo
Durante lo sviluppo del metodo, vengono selezionate le condizioni e le procedure del test che riducono al minimo l'impatto di potenziali fonti di invalidità. Venendo alla validazione statistica per an in vivo analisi, coinvolge quattro componenti principali:
- Progettazione dello studio e metodo di analisi dei dati adeguati
- Randomizzazione corretta degli animali
- Potenza statistica e dimensione del campione adeguate
- Riproducibilità adeguata tra le analisi.
Il disegno a gruppi paralleli, il disegno a blocchi randomizzati, il disegno a misure ripetute e il disegno crossover sono i tipi base di disegni sperimentali utilizzati in vivo saggio.
Di seguito sono riportati i fattori chiave che dovrebbero essere tenuti a mente durante la progettazione di un in vivo saggio:
- Tutti gli effetti biologici significativi (farmacologicamente) dovrebbero essere statisticamente significativi.
- Se non sono presenti test biologicamente rilevanti, è possibile prendere in considerazione una serie di effetti plausibili.
- Gli endpoint chiave dovrebbero essere ben definiti prima dell'inizio del test.
- Gli animali dovrebbero essere assegnati in modo casuale ai gruppi di trattamento in modo appropriato.
- I livelli di dose devono essere selezionati in modo appropriato. La selezione della dose e dell'adattamento della curva è tra gli aspetti più critici dello sviluppo del saggio biologico. La dose viene determinata in base al tipo di modello utilizzato nel segnale per adattare i dati. Per i progetti sigmoidali, un modello logistico a quattro o cinque parametri (4PL o 5PL) si adatta ai dati, mentre, per la progettazione lineare, un modello di analisi a linee parallele (PLA) si adatta ai dati.
Per un modello 4PL si consigliano nove dosi:
- Tre dosi nell'asintoto inferiore
- Tre dosi nell'asintoto superiore
- Tre dosi nell'intervallo lineare
Al contrario, per un modello PLA, si consiglia un minimo di quattro dosi. Per tracciare la curva della dose è necessario un minimo di tre dosi consecutive.
- La selezione dei gruppi di controllo e dei punti temporali per raccogliere i campioni dovrebbe essere ottimale.
- Le strategie di progettazione dovrebbero ridurre al minimo la variabilità e massimizzare le informazioni.
Comprendere la progettazione, gli sviluppi e la convalida statistica di in vivo test biologico in modo più dettagliato, contattaci all'indirizzo https://www.veedacr.com. Si possono anche leggere le linee guida menzionate da NIH visitando il link:
https://www.ncbi.nlm.nih.gov/books/NBK92013/pdf/Bookshelf_NBK92013.pdf
Riferimenti
- A. Little, "Elementi essenziali per lo sviluppo di test biologici", BioPharm International 32 (11) 2019
- Padmalayam, Ph.D., Sviluppo di test nella scoperta di farmaci
- Zwierzyna M, Overington JP (2017) Classificazione e analisi di un'ampia raccolta di descrizioni di saggi biologici in vivo. PLoS Comput Biol13(7): e1005641. https://doi.org/10.1371/journal.pcbi.1005641
- White JR, Abodeely M, Ahmed S, Debauve G, Johnson E, Meyer DM, Mozier NM, Naumer M, Pepe A, Qahwash I, Rocnik E, Smith JG, Stokes ES, Talbot JJ, Wong PY. Migliori pratiche nello sviluppo di test biologici per supportare la registrazione dei prodotti biofarmaceutici. Biotecniche. Sett. 2019;67(3):126-137. doi: 10.2144/btn-2019-0031. Epub 2019 5 agosto. PMID: 31379198.
- F Chana e Hursh D, Bioassays through the Product lifecycle: Prospettive delle revisioni CDER e CBER.
- Haas J, Manro J, Shannon H et al. Linee guida per i test in vivo. 2012 maggio 1 [Aggiornato il 2012 ottobre 1]. In: Markossian S, Grossman A, Brimacombe K, et al., a cura di. Manuale di guida al dosaggio [Internet]. Bethesda (MD): Eli Lilly & Company e il Centro nazionale per l'avanzamento delle scienze traslazionali; 2004-. URL dello scaffale: https://www.ncbi.nlm.nih.gov/books/