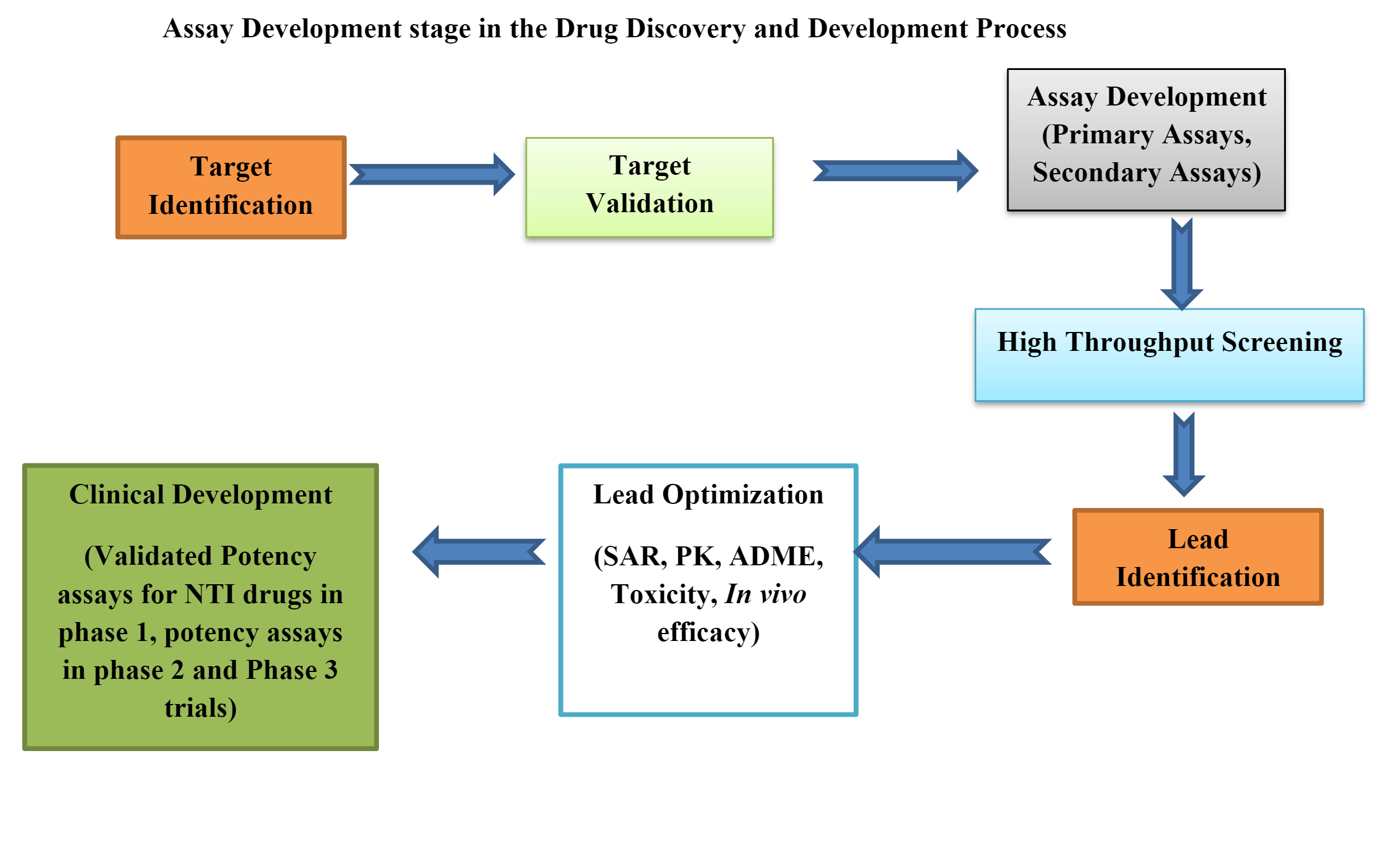
Los bioensayos participan en cada etapa del descubrimiento de fármacos, desde la identificación del objetivo hasta el descubrimiento del compuesto principal. Los bioensayos proporcionan información valiosa que muestra la potencia terapéutica de un fármaco bajo investigación.
Los datos generados durante el bioensayo también desempeñan un papel vital en el desarrollo de fármacos y el control de calidad de los productos biológicos terminados. Los bioensayos correctamente diseñados ayudan a evaluar el efecto biológico, la actividad, el proceso de transducción de señales y la capacidad de unión al receptor de un fármaco o producto biológico en un objetivo biológico (proteínas) en comparación con una referencia o estándar en un sistema biológico adecuado.
Las empresas farmacéuticas y de biotecnología involucradas en el descubrimiento y desarrollo de fármacos enfrentan continuamente el desafío de desarrollar ensayos biológicamente relevantes para el análisis de múltiples mecanismos potenciales.
El proceso implica el uso de reactivos críticos de calidad, el uso de líneas celulares específicas y fármacos de prueba purificados y productos farmacológicos de referencia que en ocasiones pueden convertirse en una limitación. La mayoría de estas actividades requieren tiempo suficiente, lo que puede convertirse en un factor limitante para los fabricantes de productos biofarmacéuticos.
Vale la pena subcontratar actividades para proveedores de servicios CRO de renombre para ahorrar tiempo en los esfuerzos de desarrollo y también para tener una opinión imparcial sobre las actividades funcionales del producto farmacéutico.
Veeda Group cuenta con científicos calificados y experimentados para diseñar, desarrollar, ejecutar y validar bioensayos para empresas y brinda servicios de bioensayos de primer nivel (in vitro y in vivo) que generan datos significativos para apoyar a las empresas farmacéuticas y de biotecnología en su viaje de descubrimiento y desarrollo de fármacos.
La experiencia de Veeda Group en el desarrollo y ejecución de bioensayos incluye:
- Prueba de neutralización por reducción de placa (ensayo PRNT)
- In Vitro Prueba de activación de línea celular humana de sensibilización cutánea (ensayo h-CLAT)
- Ensayo de captura
- Desarrollo de ensayos (farmacodinámica, farmacocinética, inmunogenicidad y evaluación de biomarcadores)
- En Vivo Bioensayos para moléculas de fármacos como hormona luteinizante, epoetina, HCG, FSH recombinante, β-HCG e insulina.
- Ensayo ADCC para biosimilares y otros ensayos diferentes como Ex vivo ensayo, ensayo basado en células, ensayo de unión a receptor, ensayo de liberación de citocinas y ensayo de ADA.
Veeda Group proporciona servicios integrados de descubrimiento, desarrollo y regulación con sus múltiples plataformas tecnológicas:
- Estudios exploratorios de toxicología.
- Estudios de toxicología regulatoria.
- In vitro Bioensayos
- Ex vivo Bioensayos
El grupo también tiene la experiencia para manejar una amplia gama de bioterapéuticos como anticuerpos monoclonales terapéuticos, insulina y análogos de insulina, citocinas, heparinas de bajo peso molecular, Biosimilares, Hormonas y biomarcadores.
El grupo Veeda ha demostrado capacidades para desarrollar proteínas recombinantes, como proteínas no glicosiladas y glicoproteínas derivadas de sistemas de expresión del huésped bacteriano o mamífero.
Bioensayos en el desarrollo de fármacos preclínicos
Los ensayos biológicos o bioensayos son herramientas esenciales en desarrollo de fármacos preclínicos. Los bioensayos preclínicos pueden ser en vivo, ex vivoy in vitro.
In vivo Los bioensayos proporcionan una medida más realista y predictiva de los efectos funcionales de las pruebas con productos farmacéuticos de referencia o material estándar de potencia definida, junto con la aplicación de herramientas estadísticas, técnicas de laboratorio específicas del estudio y el cumplimiento de un protocolo de estudio bien diseñado.
Estos ensayos capturan la complejidad del compromiso objetivo, el metabolismo y la farmacocinética de nuevos fármacos mejor que in vitro bioensayos.
Los mamíferos experimentales más utilizados en ien vivo Los ensayos de eficacia son ratones y ratas. Ocasionalmente se pueden utilizar otras especies dependiendo de la sensibilidad y la idoneidad de los ensayos.
Desarrollo y Validación de Bioensayos
Los bioensayos se utilizan como método de detección para identificar las señales que indican la actividad biológica deseada de un conjunto de compuestos. En general, un bioensayo puede generar dos tipos diferentes de señales: una dosis-respuesta lineal y una dosis-respuesta sigmoidea (en forma de S).
Dado que una solución no sirve para todos los bioensayos, es bueno evaluar y analizar los datos para desarrollar un enfoque preciso para llevar a cabo cada bioensayo.
Las etapas del ciclo de vida de un bioensayo se dividen en:
Etapa 1: Diseño, desarrollo y optimización del método.
Etapa 2: Calificación del desempeño del procedimiento
Etapa 3: Verificación del desempeño del procedimiento (adecuado para el propósito)
Desarrollar un bioensayo que cumpla con los requisitos reglamentarios y registre un producto farmacéutico es un proceso muy complejo.
El desarrollo de un bioensayo incluye muchas estrategias y diseños tácticos, como seleccionar el método correcto. in vivo plataforma, método adecuado o diseño de placa, análisis de datos, estrategia de sostenibilidad del sistema/muestra, implementación del método, desempeño del método y monitoreo.
Hay varios pasos a seguir para el desarrollo y validación de bioensayos, como la selección de dosis-respuesta y ajuste de curvas, desarrollo de referencia, cálculo de potencia, caracterización de bioensayos, diseño de calculadora de bioensayos, estandarización y automatización de bioensayos y, finalmente. , evaluación.
Tanto el desarrollo de métodos como la validación de bioensayos incluyen tres áreas fundamentales:
- Validación previa al estudio (fase de identificación y diseño)
- Validación en estudio (fase de desarrollo y producción)
- Validación cruzada o validación de transferencia de método
Durante el desarrollo del método, se seleccionan condiciones y procedimientos de ensayo que minimicen el impacto de posibles fuentes de invalidez. Llegando a la validación estadística de un in vivo ensayo, consta de cuatro componentes principales:
- Diseño de estudio y método de análisis de datos adecuados.
- Aleatorización adecuada de los animales.
- Poder estadístico y tamaño de muestra adecuados.
- Reproducibilidad adecuada entre ejecuciones de ensayo.
El diseño de grupos paralelos, el diseño de bloques aleatorios, el diseño de medidas repetidas y el diseño cruzado son los tipos básicos de diseños experimentales utilizados en in vivo ensayo.
Los siguientes son los factores clave que se deben tener en cuenta al diseñar un in vivo ensayo:
- Todos los efectos biológicos significativos (farmacológicamente) deben ser estadísticamente significativos.
- Si no existen ensayos biológicamente relevantes, entonces se puede considerar una variedad de efectos plausibles.
- Los criterios de valoración clave deben estar bien definidos antes del comienzo del ensayo.
- Los animales deben asignarse aleatoriamente de manera adecuada a los grupos de tratamiento.
- Los niveles de dosis deben seleccionarse apropiadamente. La selección de dosis y ajuste de curvas se encuentra entre los aspectos más críticos del desarrollo de bioensayos. La dosis se determina dependiendo del tipo de modelo utilizado en la señal para ajustar los datos. Para los diseños sigmoidales, un modelo logístico de cuatro o cinco parámetros (4PL o 5PL) se ajusta a los datos, mientras que, para el diseño lineal, un modelo de análisis de líneas paralelas (PLA) se ajusta a los datos.
Para un modelo 4PL se recomiendan nueve dosis:
- Tres dosis en la asíntota inferior
- Tres dosis en la asíntota superior
- Tres dosis en el rango lineal
En cambio, para un modelo de PLA se recomienda un mínimo de cuatro dosis. Se requiere un mínimo de tres dosis consecutivas para trazar la curva de dosis.
- La selección de grupos de control y momentos para recolectar muestras debe ser óptima.
- Las estrategias de diseño deben minimizar la variabilidad y maximizar la información.
Comprender el diseño, desarrollo y validación estadística de in vivo bioensayo con más detalle, comuníquese con nosotros en https://www.veedacr.com. También se pueden leer las pautas mencionadas por los NIH visitando el enlace:
https://www.ncbi.nlm.nih.gov/books/NBK92013/pdf/Bookshelf_NBK92013.pdf
Referencias
- A. Little, “Fundamentos esenciales en el desarrollo de bioensayos”, BioPharm International 32 (11) 2019
- Padmalayam, Ph.D., Desarrollo de ensayos en el descubrimiento de fármacos
- Zwierzyna M, Overington JP (2017) Clasificación y análisis de una gran colección de descripciones de bioensayos in vivo. PLoS Comput Biol13(7): e1005641. https://doi.org/10.1371/journal.pcbi.1005641
- White JR, Abodeely M, Ahmed S, Debauve G, Johnson E, Meyer DM, Mozier NM, Naumer M, Pepe A, Qahwash I, Rocnik E, Smith JG, Stokes ES, Talbot JJ, Wong PY. Mejores prácticas en el desarrollo de bioensayos para apoyar el registro de productos biofarmacéuticos. Biotecnicas. 2019 septiembre;67(3):126-137. doi: 10.2144/btn-2019-0031. Publicación electrónica del 2019 de agosto de 5. PMID: 31379198.
- F Chana y Hursh D, Bioensayos a lo largo del ciclo de vida del producto: perspectivas de las revisiones CDER y CBER.
- Haas J, Manro J, Shannon H, et al. Directrices para ensayos in vivo. 2012 de mayo de 1 [Actualizado el 2012 de octubre de 1]. En: Markossian S, Grossman A, Brimacombe K, et al., editores. Manual de orientación de ensayos [Internet]. Bethesda (MD): Eli Lilly & Company y el Centro Nacional para el Avance de las Ciencias Traslacionales; 2004-. URL de la estantería: https://www.ncbi.nlm.nih.gov/books/