
Indien håller på att växa fram som ett land med en enorm potential att bidra till de nationella och internationella plattformarna för kliniska prövningar.
Smakämnen Central Drugs Standard Control Organisation (CDSCO) är Indiens nationella tillsynsmyndighet som hanteras av Drug Controller General of India (DCGI).1,2,3
DCGI ansvarar för samordningen av inspektioner av sponsorer, tillverkningsenheter, såväl som spårplatser.3
Tidiga år i klinisk utveckling
År 2000 upprättade Indian Council of Medical Research (ICMR) etiska riktlinjer för att bedriva biomedicinsk forskning på mänskliga ämnen.4 År 2005 sågs en översyn av schema Y i Drug & Cosmetics Act, 1945, för att anpassa indiska lagar till definitioner och förfaranden som är internationellt accepterade.
Ändringarna inkluderade:
- Definiera Fas I till fas IV av en studie
- Avgränsat ansvar för sponsor(er) och utredare(r)
- Alternativ för att registrera eventuella avvikelser eller ändringar i det godkända studieprotokollet
Indien undertecknade också avtalet om handelsrelaterade immateriella rättigheter (TRIPS) 2005 för att öppna upp möjligheterna att genomföra fler kliniska prövningar i Indien.5
Bortsett från att harmonisera lagstiftningen till internationella standarder blev Indien snabbt en gynnsam destination för kliniska prövningar eftersom det erbjöd:6
- Engelsktalande yrkesverksamma inom hälso- och sjukvården
- Teknisk expertis
- Växande ekonomi
- Teknik i världsklass
- Stor, mångsidig och behandlingsnaiv befolkning
Ställ tillbaka för kliniska prövningar
Trots ändringar i regelverket utnyttjade många multinationella läkemedelsföretag den stora befolkning som antingen hade otillräcklig kunskap om kliniska prövningar eller var analfabeter. Dessutom bidrog ett dåligt definierat sjukvårdssystem till utmaningarna med att övervaka oetiska metoder.
Detta ledde till att kliniska prövningar genomfördes med liten övervakning och ingen inspelning av patientens informerade samtycke, vare sig skriftligt eller som audio/visuellt innehåll.
Patienterna administrerades prövningsläkemedel eller anordningar utan att avslöja kända allvarliga biverkningar, några ledde till försökspersonernas död. Dessutom inrättades ingen oberoende utredningskommitté för att fastställa om patientens död var relaterad/inte relaterad till undersökningsprodukten eller produkten.4
Åren 2010 till 2013 såg en prövande fas i Indisk klinisk prövning scenario på grund av de ackumulerade negativa effekterna av att genomföra oetiska prövningar.
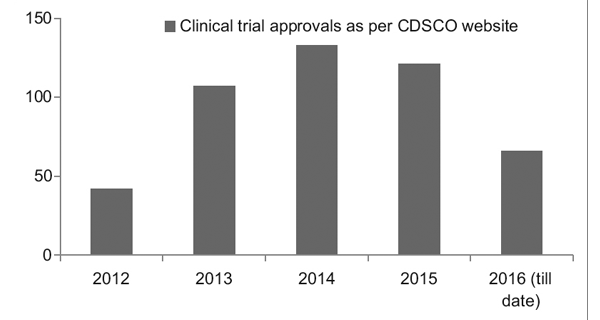
Men med ett bättre regelverk på plats har Clinical Trial Registry of India (CTRI) registrerat en stadig ökning av antalet prövningar som genomförs, som ses i figur 1. Det observerades också att de flesta prövningarna var fas III prövningar.7
Figur 1: Kliniska prövningstrender genom åren
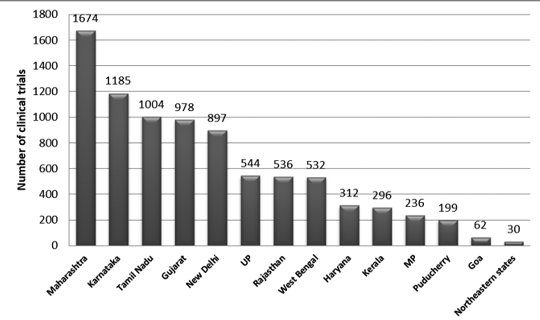
Figur 2 visar den statliga fördelningen av försök i Indien mellan 2007 och 2015. Cirka 3330 spår registrerades under denna period.
Det observerades att det maximala antalet försök genomfördes i Maharashtra, och det minsta antalet försök utfördes i den nordöstra delstaten. Bland de nordöstra delstaterna genomfördes inga rättegångar i Nagaland.7
Figur 2: Statlig fördelning av kliniska prövningar i Indien (2007-2015 data)7
Återupplivande av det kliniska och regulatoriska scenariot
Under 2014 bildade CDSCO 12 nya läkemedelsrådgivande kommittéer (NDAC) och 25 ämnesexpertkommittéer (SECs). Dessa kommittéer har ett antal experter från framstående statliga högskolor och institutioner för att påskynda godkännandetidslinjerna för en klinisk prövning till 6-7 månader.
Processen i tre nivåer består av:9
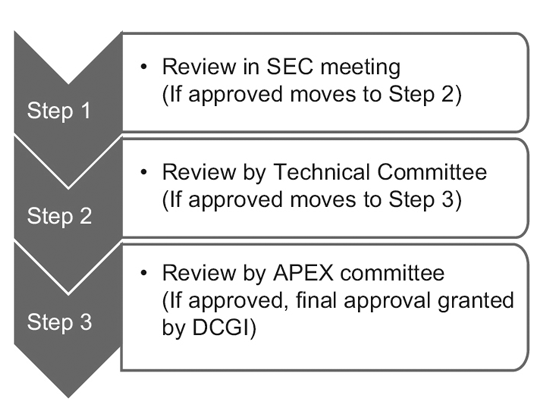
Det är dock endast SEC som granskar globala ansökningar om kliniska prövningar, och inget ytterligare godkännande krävs från den tekniska kommittén eller Apex-kommittén. Ansökningar om nya läkemedel (IND) granskas också oberoende av IND-kommittén och kräver inte godkännande av Apex-kommittén.
En teknisk kommitté kommer in i bilden endast om SEC har avslagit en sponsors ansökan och sponsorn känner sig förolämpad över beslutet. I ett sådant fall, om den tekniska kommittén inte håller med SEC:s beslut, har den befogenhet att åsidosätta SEC:s beslut.10
I mars 2019 släppte ministeriet för hälsa och familjeskydd i Indien Nya läkemedel och regler för kliniska prövningar 2019 med avsikten att påskynda godkännandet av kliniska prövningar, nya läkemedel, bioekvivalens (BE) eller biotillgänglighetsstudier (BA).
Dessa regler har också tagit itu med eventuella oklarheter som funnits med avseende på regleringen av den etiska kommittén (EC).11
Höjdpunkter i de nya reglerna för droger och kliniska prövningar, 201911 |
Uppdaterade regler och föreskrifter11 |
Tidslinje för godkännande av kliniska prövningar |
90 arbetsdagar från mottagandet av en ansökan om läkemedel som upptäckts utanför Indien och 30 arbetsdagar för nya läkemedel eller IND i Indien |
Tillverkning av nya läkemedel eller IND, BE & BA studier eller testanalys eller undersökning |
Tillstånd krävs från Central Licensing Authority (CLA) |
Avstående från lokala kliniska prövningar |
· Om CLA har godkänt marknadsföringen av det nya läkemedlet i andra länder eller har gett tillstånd att genomföra globala kliniska prövningar för det nya läkemedlet i Indien · Inga bevis för en skillnad i metabolism, säkerhet eller effekt på grund av skillnaden i den indiska befolkningens genetiska profil |
Giltighetstid för en klinisk prövning |
2 år från datumet för utfärdandet av CLA |
Tillgång efter prövning till IND eller nytt läkemedel |
Under unika omständigheter ska läkemedlet distribueras kostnadsfritt till försökspersoner enligt CLA:s anvisningar, men inget ansvar ligger på sponsorn för användningen av läkemedlet efter prövningen. |
Möten före och efter inlämning |
Att söka vägledning med avseende på lagar och procedurer som styr processen för tillverkning och licensiering eller beviljande av tillstånd. |
Godkännande för prövningar utförda av EC och registrering av EC |
· Godkännande ska erhållas från EC för en annan prövningsplats om en prövningsplats inte har en EC, och EC bör befinna sig inom 50 km från prövningsplatsen.· CLA-godkänd registrering av EC förblir giltig i fem år från utfärdandedatum. |
Villkor som ska uppfyllas för att genomföra en klinisk prövning |
· Inlämning av statusrapport på kvartalsbasis eller beroende på försökets längd för att spåra ämnesregistrering· Onlinerapportering av status för den kliniska prövningen var sjätte månad via SUGHAM-portalen för att veta om prövningen pågår eller är avslutad eller har avslutats. |
Avgift för anskaffning av licens, registreringsbevis och tillstånd för prövning |
Olika avgiftsstrukturer beroende på syftet med försöket. Avgift från INR 50,000 5,00,000 till XNUMX XNUMX XNUMX. |
Att överbrygga klyftan
Utmaningarna med att hantera kliniska prövningar är mångfacetterade och involverar att följa regelverket på ett ansvarsfullt och etiskt sätt av såväl intressenter, myndigheter som rättssystem.
Patientsäkerhet och skydd bör vara av yttersta vikt att sätta strikta regler för:12
- informerat samtycke genom audiovisuell inspelning och på ett språk som patienten är bekväm med
- respekt för patientens kulturella, sociala, ekonomiska och utbildningsmässiga bakgrund
- snabb rapportering av SAE
Nya spelregler som kan öppna upp möjligheten att utöka medicinsk forskning i Indien är:13
- godkännande av förslag som lämnats in till DCGI inom 30 dagar efter ansökan, om det inte finns någon kommunikation från DCGI
- snabb spårning av inhemska godkännanden
- möten före och efter inlämning med expertkommittén för att få in mer insyn i processen och för att fastställa en väldefinierad tidslinje för slutförandet av provet
- rättegångsersättning om det prövningsläkemedlet ledde till SAE/död.
Skicklig arbetskraft och toppmodern infrastruktur spelar också en viktig roll för att attrahera sponsorföretag. Forskning har visat att även om fas III-studier genomförs i stor utsträckning i Indien, verkar fas I-studier vara begränsade till sponsorlandet.
Detta kan tillskrivas sponsorns oro för att skaffa kvalificerad arbetskraft och teknik. För att möjliggöra forskning om ursprungsbefolkningen i Indien är det avgörande att tillhandahålla lämplig exponering eller fortlöpande medicinsk utbildning till personal och tillgång till uppdaterad teknologi för att erkännas som ett land som är kompetent nog att genomföra alla fasförsök.9
Lika viktigt är behovet av att kvalificerad vårdpersonal finns tillgänglig i hela landet för att ta hänsyn till den ojämna fördelningen av kliniska prövningar mellan stater.
Att koncentrera en prövning på ett visst tillstånd kan leda till partiska slutsatser och förenkla eller överdriva en sjukdomsbörda eller tillstånd. Genom att ge tillgång till människor i alla stater att gå med i en klinisk prövning, minimerar vi inte bara partiskhet utan inkluderar också olika etniska befolkningsgrupper.9
Framtiden
Med positiva, patientvänliga, snabba och transparenta regulatoriska lagar kommer Indien att fortsätta växa som ett internationellt nav för att testa och utveckla innovativa läkemedel och medicintekniska produkter.
Källor
1. Evangeline L, Mounica NVN, Reddy VS et al,. Regulatorisk process och etik för kliniska prövningar i Indien (CDSCO). The Pharma Innovation Journal. 2017;6(4):165-9. http://www.thepharmajournal.com/archives/2017/vol6issue4/PartC/6-4-4-176.pdf
2. Lahiry S, Sinha R, Choudhary S et al,. Paradigmskifte i bestämmelser om kliniska prövningar i Indien. Indian Journal of Reumatology. 2018; 13: 51-5.
3. Gogtay NJ, Ravi R och Thatte UM. Regulatoriska krav för kliniska prövningar i Indien: Vad akademiker behöver veta. Indian Journal of Anesthesia. 2017 Mar;61(3):192-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5372399/
4. Ramu B, Kumar MS och Ramakrishna N. Aktuellt regulatoriskt scenario för genomförande av kliniska prövningar i Indien. Pharmaceutical Regulatory Affairs. Fri tillgång. 2015; 4: 2. https://www.researchgate.net/publication/281765214_Current_Regulatory_Scenario_for_Conducting_Clinical_Trials_in_India
5. Burt T, Sharma P, Dhillon S et al,. Klinisk forskningsmiljö i Indien: utmaningar och föreslagna lösningar. Journal of Clinical Research Bioethics. 2014;5:6. DOI: 10.4172/2155-9627.1000201
6. Chaturvedi M, Gogtay NJ, Thatte UM. Matchar kliniska prövningar i Indien dess behov av sjukvård? En revision av Clinical Trials Registry of India. Perspektiv i klinisk forskning. 2017;8(4):172-5.
7. http://ctri.nic.in/Clinicaltrials/news/CTRI_Newsbulletin_July-Dec_2017.pdf Öppnas den 23 april 2019.
8. Bhave A och Menon S. Regulatorisk miljö för klinisk forskning: Senaste förflutna och förväntad framtid. Perspektiv i klinisk forskning. 2017; 8: 11.6.
9. Viktiga höjdpunkter i nya droger och regler för kliniska prövningar, 2019. Tillträde den 23 april 2019
10. Dan S, Karmakar S, Ghosh B et al,. Digitalisering av kliniska prövningar i Indien: ett nytt steg av CDSCO mot att säkerställa datatrovärdighet och patientsäkerhet. Pharmaceutical Regulatory Affairs: Open Access. 2015;4(3): DOI: 10.4172/2167-7689.1000149.
11. https://www.thehindubusinessline.com/news/new-rules-sweeten-the-deal-for-clinical-trials-by-indian-pharma-cos/article26283499.ece Åtkomst den 23 april 2019.
Varning:
Informationen i denna artikel är endast avsedd att ge allmän vägledning i frågor av intresse för personligt bruk av läsaren, som tar fullt ansvar för dess användning. Följaktligen tillhandahålls informationen i denna artikel under förutsättning att författaren/författarna och utgivaren/utgivarna inte här är engagerade i att tillhandahålla professionella råd eller tjänster.
Som sådan bör den inte användas som ett substitut för konsultation med en kompetent rådgivare. Innan du fattar något beslut eller vidtar någon åtgärd bör läsaren alltid rådfråga en professionell rådgivare angående den relevanta artikelinlägget.
Även om alla försök har gjorts för att säkerställa att informationen i den här artikeln har erhållits från tillförlitliga källor, är Veeda Clinical Research inte ansvarig för några fel eller utelämnanden eller för de resultat som erhållits från användningen av denna information.
All information i den här artikeln tillhandahålls "i befintligt skick" utan garanti för fullständighet, exakthet, aktualitet eller för de resultat som erhålls från användningen av denna information, och utan garantier av något slag, uttryckliga eller underförstådda, inklusive men inte begränsat till garantier för prestanda, säljbarhet och lämplighet för ett visst ändamål.
Ingenting häri ska i någon utsträckning ersätta de oberoende undersökningarna och läsarens sunda tekniska och affärsmässiga bedömning. Under inga omständigheter kommer Veeda klinisk forskning, eller dess partners, anställda eller agenter, vara ansvariga gentemot läsaren eller någon annan för alla beslut som fattas eller åtgärder som vidtas med stöd av informationen i denna artikel eller för eventuella följdskador, speciella eller liknande skador, även om de informeras om möjligheten. av sådana skador.
Ingen del av denna publikation får reproduceras, lagras i ett hämtningssystem eller överföras i någon form eller på något sätt, mekaniskt, elektroniskt, genom fotokopiering, inspelning eller på annat sätt, utan föregående skriftligt tillstånd från utgivaren.
För information, kontakta oss på:
Veeda Clinical Research Private Limited
Vedant Complex, Beside YMCA Club, SG Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat Indien.
Telefon: + 91-79-3001-3000
Fax: + 91-79-3001-3010
e-post: info@veedacr.com


{kind=link}