
L’India sta emergendo come un paese con un enorme potenziale per contribuire alle piattaforme di sperimentazione clinica nazionali e internazionali.
I Organizzazione centrale per il controllo degli standard dei farmaci (CDSCO) è l'autorità nazionale di regolamentazione dell'India gestita dal Drug Controller General of India (DCGI).1,2,3
Il DCGI è responsabile del coordinamento delle ispezioni degli sponsor, delle unità produttive e dei siti di tracciamento.3
Primi anni di sviluppo clinico
Nel 2000, l’Indian Council of Medical Research (ICMR) ha stabilito linee guida etiche per la conduzione della ricerca biomedica su soggetti umani.4 Il 2005 ha visto una revisione della Tabella Y del Drug & Cosmetics Act del 1945, per allineare le leggi normative indiane alle definizioni e alle procedure accettate a livello internazionale.
Le modifiche includevano:
- Definizione Fase I alla Fase IV di uno studio
- Responsabilità delimitate dello sponsor e dello sperimentatore
- Opzioni per registrare qualsiasi deviazione o modifica al protocollo di studio approvato
L'India ha anche firmato nel 2005 l'accordo sui diritti di proprietà intellettuale legati al commercio (TRIPS) al fine di aprire le prospettive per la conduzione di più studi clinici in India.5
Oltre ad armonizzare gli atti normativi agli standard internazionali, l’India è diventata rapidamente una destinazione favorevole per le sperimentazioni cliniche poiché offriva:6
- Professionisti di lingua inglese nel settore sanitario
- Competenza tecnica
- Economia in crescita
- Tecnologia di prim'ordine
- Popolazione ampia, diversificata e naïve al trattamento
Arretrato per gli studi clinici
Nonostante i cambiamenti nelle normative, molte aziende farmaceutiche multinazionali hanno approfittato dell’ampia popolazione che aveva una conoscenza inadeguata sugli studi clinici o era analfabeta. Inoltre, un sistema sanitario mal definito si aggiungeva alle sfide legate al monitoraggio delle pratiche non etiche.
Ciò ha portato a condurre studi clinici con scarsa supervisione e senza alcuna registrazione del consenso informato del paziente né in forma scritta né come contenuto audio/visivo.
Ai pazienti sono stati somministrati farmaci o dispositivi sperimentali senza rivelare effetti avversi gravi noti, alcuni dei quali hanno portato alla morte dei soggetti. Inoltre, non è stato istituito alcun comitato d'inchiesta indipendente per accertare se la morte del paziente fosse correlata/non correlata al prodotto o dispositivo in sperimentazione.4
Gli anni dal 2010 al 2013 hanno visto una fase difficile nel Studio clinico indiano scenario a causa degli effetti negativi accumulati derivanti dalla conduzione di processi non etici.
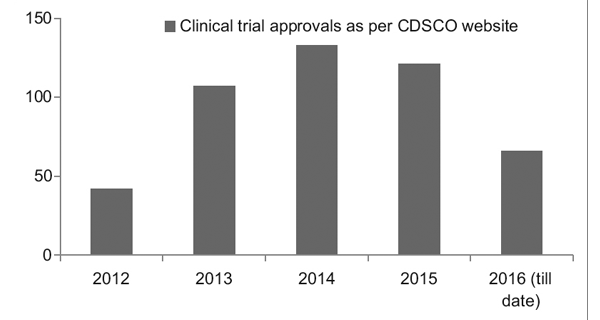
Tuttavia, con un migliore quadro normativo in atto, il Clinical Trial Registry of India (CTRI) ha registrato un aumento costante nel numero di studi condotti, come mostrato nella Figura 1. È stato inoltre osservato che la maggior parte degli studi erano di fase III prove.7
figura 1: Andamento degli studi clinici nel corso degli anni
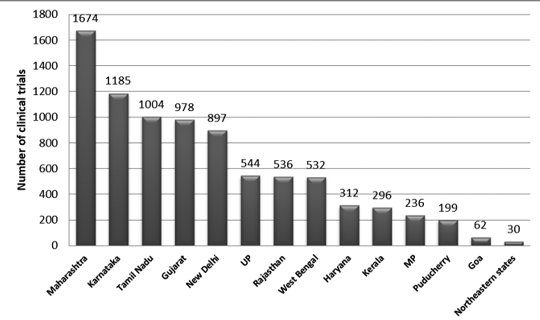
La Figura 2 presenta la distribuzione a livello statale delle sperimentazioni in India tra il 2007 e il 2015. Durante questo periodo sono state registrate circa 3330 tracce.
È stato osservato che il numero massimo di prove è stato condotto nel Maharashtra e il minor numero di prove è stato condotto nello stato nordorientale. Tra gli stati del nord-est, nel Nagaland non è stato condotto alcun processo.7
figura 2: Distribuzione a livello statale degli studi clinici in India (dati 2007-2015)7
Rilancio dello scenario clinico e regolatorio
Nel 2014, il CDSCO ha costituito 12 nuovi comitati consultivi sui farmaci (NDAC) e 25 comitati di esperti in materia (SEC). Questi comitati comprendono un numero di esperti provenienti da eminenti college e istituzioni governative per accelerare i tempi di approvazione di una sperimentazione clinica a 6-7 mesi.
Il processo a tre livelli è composto da:9
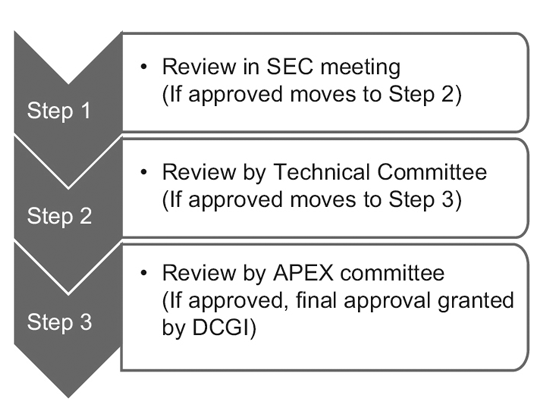
Tuttavia, solo la SEC esamina le domande di sperimentazione clinica globale e non è richiesta alcuna ulteriore approvazione da parte del comitato tecnico o del comitato Apex. Anche le richieste di sperimentazione di nuovi farmaci (IND) vengono esaminate in modo indipendente dal comitato IND e non richiedono l'approvazione del comitato Apex.
Un comitato tecnico entra in gioco solo se la SEC ha respinto la richiesta di uno sponsor e lo sponsor si sente leso dalla decisione. In tal caso, se il comitato tecnico non è d'accordo con la decisione della SEC, ha il potere di annullare la decisione della SEC.10
Nel marzo 2019, il Ministero della salute e del benessere familiare dell’India ha rilasciato il Nuovi farmaci e norme sulla sperimentazione clinica 2019 con l’intenzione di accelerare l’approvazione di sperimentazioni cliniche, nuovi farmaci, studi di bioequivalenza (BE) o di biodisponibilità (BA).
Queste regole hanno anche affrontato ogni ambiguità esistente rispetto al regolamento del Comitato Etico (CE).11
Punti salienti dei nuovi farmaci e delle norme sulla sperimentazione clinica, 201911 |
Norme e regolamenti aggiornati11 |
Tempistiche di approvazione per gli studi clinici |
90 giorni lavorativi dal ricevimento di una domanda per farmaci scoperti al di fuori dell'India e 30 giorni lavorativi per nuovi farmaci o IND in India |
Produzione di nuovi farmaci o studi IND, BE e BA o analisi o esami di test |
È necessaria l'autorizzazione della Central Licensing Authority (CLA) |
Rinuncia agli studi clinici locali |
· Se il CLA ha approvato la commercializzazione del nuovo farmaco in altri paesi o ha concesso il permesso di condurre studi clinici globali per il nuovo farmaco in India · Nessuna prova di differenza nel metabolismo, nella sicurezza o nell'efficacia a causa della differenza nel profilo genetico della popolazione indiana |
Periodo di validità di una sperimentazione clinica |
2 anni dalla data di rilascio da parte del CLA |
Accesso post-esperimento all'IND o al nuovo farmaco |
In circostanze uniche, il farmaco deve essere distribuito gratuitamente ai soggetti dello studio secondo le indicazioni del CLA, ma nessuna responsabilità spetta allo sponsor per l'uso del farmaco dopo lo studio. |
Incontri pre-presentazione e post-presentazione |
Cercare assistenza rispetto alla legge e alle procedure che regolano il processo di produzione e concessione di licenze o permessi. |
Approvazione per le sperimentazioni condotte dalla CE e registrazione della CE |
· Occorre ottenere l'approvazione da parte della CE di un altro sito di sperimentazione se un sito di sperimentazione non ha un EC e la CE deve trovarsi entro 50 km dal sito di sperimentazione.· La registrazione CE approvata dal CLA rimane valida per cinque anni dalla data di rilascio. |
Condizioni da soddisfare per la conduzione di una sperimentazione clinica |
· Invio del rapporto sullo stato su base trimestrale o in base alla durata dello studio per tenere traccia dell'iscrizione del soggetto· Reporting online dello stato della sperimentazione clinica ogni sei mesi tramite il portale SUGHAM per sapere se la sperimentazione è in corso, completata o terminata. |
Tassa per l'ottenimento di una licenza, certificato di registrazione e permesso di prova |
Differenti strutture tariffarie a seconda dello scopo della sperimentazione. La tariffa varia da INR 50,000 a INR 5,00,000. |
Colmare il divario
Le sfide legate alla gestione degli studi clinici sono molteplici e implicano il rispetto del quadro normativo in modo responsabile ed etico da parte delle parti interessate, del governo e del sistema giudiziario.
Sicurezza e tutela del paziente dovrebbe essere della massima importanza stabilire norme rigorose per:12
- consenso informato mediante registrazione audiovisiva e in una lingua con cui il paziente si sente a suo agio
- rispetto del contesto culturale, sociale, economico ed educativo del paziente
- segnalazione tempestiva dei SAE
Nuove regole di base che possono aprire la possibilità di espandere la ricerca medica in India sono:13
- approvazione delle proposte presentate alla DCGI entro 30 giorni dalla domanda, in assenza di comunicazione da parte della DCGI
- monitoraggio rapido delle approvazioni nazionali
- riunioni pre e post-presentazione con il comitato di esperti per garantire maggiore trasparenza al processo e stabilire una tempistica ben definita per il completamento della sperimentazione
- risarcimento del processo nel caso in cui il farmaco sperimentale abbia portato a SAE/morte.
Forza lavoro competente e infrastrutture all'avanguardia svolgono un ruolo importante anche nell’attrarre aziende sponsor. La ricerca ha dimostrato che, sebbene gli studi di Fase III siano condotti su larga scala in India, gli studi di Fase I sembrano essere limitati al paese sponsor.
Ciò potrebbe essere attribuito alla preoccupazione dello Sponsor nel procurarsi forza lavoro e tecnologia qualificate. Per consentire la ricerca indigena in India, è fondamentale fornire un’adeguata esposizione o formazione medica continua al personale e l’accesso a tecnologie aggiornate per essere riconosciuto come un paese sufficientemente competente per condurre qualsiasi sperimentazione di fase.9
Altrettanto importante è la necessità che operatori sanitari qualificati siano disponibili in tutto il Paese per tenere conto della distribuzione non uniforme degli studi clinici tra gli Stati.
Concentrare uno studio su un particolare stato potrebbe portare a conclusioni distorte e semplificare eccessivamente o esagerare il carico o la condizione della malattia. Fornendo l'accesso a persone di tutti gli stati per partecipare a una sperimentazione clinica, non solo riduciamo al minimo i pregiudizi, ma includiamo anche diverse popolazioni etniche.9
Il futuro
Grazie a leggi normative positive, favorevoli ai pazienti, rapide e trasparenti, l’India continuerà a crescere come hub internazionale per testare e sviluppare farmaci e dispositivi medici innovativi.
fonti
1. Evangeline L, Mounica NVN, Reddy VS et al. Processo normativo ed etica per gli studi clinici in India (CDSCO). Il giornale dell'innovazione farmaceutica. 2017;6(4):165-9. http://www.thepharmajournal.com/archives/2017/vol6issue4/PartC/6-4-4-176.pdf
2. Lahiry S, Sinha R, Choudhary S et al. Cambiamento di paradigma nella regolamentazione degli studi clinici in India. Giornale indiano di reumatologia. 2018; 13: 51-5.
3. Gogtay NJ, Ravi R e Thatte UM. Requisiti normativi per le sperimentazioni cliniche in India: cosa devono sapere gli accademici. Giornale indiano di anestesia. 2017 Mar;61(3):192-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5372399/
4. Ramu B, Kumar MS e Ramakrishna N. Scenario normativo attuale per la conduzione di studi clinici in India. Affari regolatori farmaceutici. Accesso libero. 2015; 4: 2. https://www.researchgate.net/publication/281765214_Current_Regulatory_Scenario_for_Conducting_Clinical_Trials_in_India
5. Burt T, Sharma P, Dhillon S et al. Ambiente di ricerca clinica in India: sfide e soluzioni proposte. Giornale di bioetica della ricerca clinica. 2014;5:6. DOI: 10.4172/2155-9627.1000201
6. Chaturvedi M, Gogtay NJ, Thatte UM. Gli studi clinici condotti in India soddisfano le sue esigenze sanitarie? Un audit del registro degli studi clinici dell’India. Prospettive nella ricerca clinica. 2017;8(4):172-5.
7. http://ctri.nic.in/Clinicaltrials/news/CTRI_Newsbulletin_July-Dec_2017.pdf Accesso effettuato il 23 aprile 2019.
8. Bhave A e Menon S. Ambiente normativo per la ricerca clinica: passato recente e futuro atteso. Prospettive nella ricerca clinica. 2017; 8: 11.6.
9. Punti salienti dei nuovi farmaci e delle norme sulla sperimentazione clinica, 2019. Accesso effettuato il 23 aprile 2019
10. Dan S, Karmakar S, Ghosh B et al. Digitalizzazione degli studi clinici in India: un nuovo passo di CDSCO verso la garanzia della credibilità dei dati e della sicurezza dei pazienti. Affari di regolamentazione farmaceutica: accesso aperto. 2015;4(3): DOI: 10.4172/2167-7689.1000149.
11 https://www.thehindubusinessline.com/news/new-rules-sweeten-the-deal-for-clinical-trials-by-indian-pharma-cos/article26283499.ece Accesso effettuato il 23 aprile 2019.
Disclaimer:
Le informazioni contenute in questo articolo hanno lo scopo esclusivo di fornire indicazioni generali su questioni di interesse per l'uso personale del lettore, che si assume la piena responsabilità del loro utilizzo. Di conseguenza, le informazioni contenute in questo articolo sono fornite con la consapevolezza che l'autore(i) e l'editore(i) non sono qui impegnati a fornire consulenza o servizi professionali.
Pertanto, non deve essere utilizzato in sostituzione della consultazione di un consulente competente. Prima di prendere qualsiasi decisione o intraprendere qualsiasi azione, il lettore dovrebbe sempre consultare un consulente professionale relativo all'articolo in questione.
Sebbene sia stato fatto ogni sforzo per garantire che le informazioni contenute in questo articolo siano state ottenute da fonti affidabili, Veeda Clinical Research non è responsabile per eventuali errori o omissioni o per i risultati ottenuti dall'uso di queste informazioni.
Tutte le informazioni contenute in questo articolo sono fornite "così come sono", senza alcuna garanzia di completezza, accuratezza, tempestività o dei risultati ottenuti dall'uso di tali informazioni e senza garanzia di alcun tipo, espressa o implicita, incluso, ma non limitato alle garanzie di prestazione, commerciabilità e idoneità per uno scopo particolare.
Nulla di quanto contenuto nel presente documento potrà, in alcun modo, sostituire le indagini indipendenti e il valido giudizio tecnico e commerciale del lettore. In nessun caso lo farà Ricerca clinica Veeda, o i suoi partner, dipendenti o agenti, sono responsabili nei confronti del lettore o di chiunque altro per qualsiasi decisione presa o azione intrapresa facendo affidamento sulle informazioni contenute in questo articolo o per eventuali danni consequenziali, speciali o simili, anche se informati della possibilità di tali danni.
Nessuna parte di questa pubblicazione può essere riprodotta, archiviata in un sistema di recupero o trasmessa in qualsiasi forma o con qualsiasi mezzo, meccanico, elettronico, fotocopiatrice, registrazione o altro, senza il previo consenso scritto dell'editore.
Per informazioni contattateci a:
Veeda Clinical Research Private Limited
Complesso Vedant, accanto al YMCA Club, SG Highway,
Vejalpur, Ahmedabad – 380 051,
Gujarat India.
Telefono: + 91-79-3001-3000
Fax: + 91-79-3001-3010
E-mail: info@veedacr.com


{kind=link}