
India está emergiendo como un país con un enorme potencial para contribuir a las plataformas de ensayos clínicos nacionales e internacionales.
El Organización Central de Control de Estándares de Drogas (CDSCO) es la autoridad reguladora nacional de la India gestionada por el Contralor General de Medicamentos de la India (DCGI).1,2,3
La DCGI es responsable de la coordinación de las inspecciones de los patrocinadores, las unidades de fabricación y los sitios de los senderos.3
Primeros años en el desarrollo clínico
En 2000, el Consejo Indio de Investigación Médica (ICMR) estableció directrices éticas para realizar investigaciones biomédicas en seres humanos.4 En el año 2005 se realizó una revisión del Anexo Y de la Ley de Medicamentos y Cosméticos de 1945, para alinear las leyes regulatorias indias con las definiciones y procedimientos aceptados internacionalmente.
Los cambios incluyeron:
- Definición fase I a la Fase IV de un ensayo
- Responsabilidades demarcadas de los patrocinadores y los investigadores
- Opciones para registrar cualquier desviación o cambio en el protocolo de estudio aprobado.
India también firmó el acuerdo sobre derechos de propiedad intelectual relacionados con el comercio (ADPIC) en 2005 con el fin de abrir perspectivas para realizar más ensayos clínicos en India.5
Además de armonizar las leyes regulatorias con los estándares internacionales, la India rápidamente se convirtió en un destino favorable para los ensayos clínicos, ya que ofrecía:6
- Profesionales de habla inglesa en el ámbito de la salud.
- Conocimientos técnicos
- Economía en crecimiento
- Tecnología de clase mundial
- Población numerosa, diversa y sin tratamiento previo
Retraso para ensayos clínicos
A pesar de los cambios en las regulaciones, muchas compañías farmacéuticas multinacionales se aprovecharon de la gran población que tenía conocimientos inadecuados sobre los ensayos clínicos o era analfabeta. Además, un sistema de salud mal definido se sumó a los desafíos de monitorear las prácticas poco éticas.
Esto llevó a realizar ensayos clínicos con poca supervisión y sin registrar el consentimiento informado del paciente, ya sea por escrito o como contenido audiovisual.
A los pacientes se les administraron medicamentos o dispositivos en investigación sin revelar efectos adversos graves conocidos, algunos de los cuales provocaron la muerte de los sujetos. Además, no se creó ningún comité de investigación independiente para determinar si la muerte del paciente estaba relacionada o no con el producto o dispositivo en investigación.4
Los años 2010 a 2013 vieron una fase difícil en el ensayo clínico indio escenario debido a los efectos nocivos acumulados de la realización de juicios poco éticos.
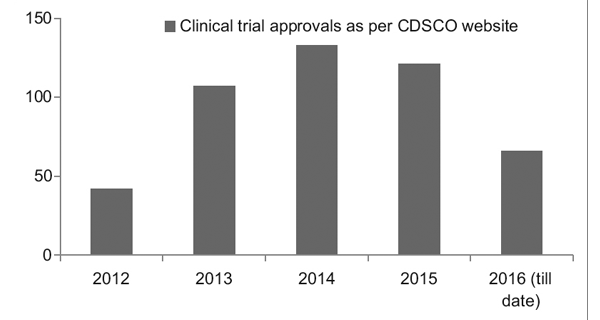
Sin embargo, con un mejor marco regulatorio, el Registro de Ensayos Clínicos de la India (CTRI) ha registrado un aumento constante en el número de ensayos que se llevan a cabo, como se ve en la Figura 1. También se observó que la mayoría de los ensayos eran de fase III. ensayos.7
Figura 1: Tendencias de los ensayos clínicos a lo largo de los años
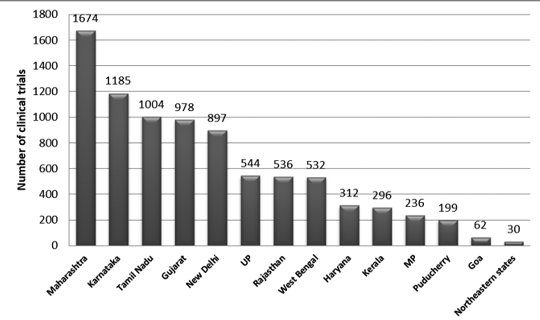
La Figura 2 presenta la distribución estatal de ensayos en la India entre 2007 y 2015. Durante este período se registraron aproximadamente 3330 senderos.
Se observó que el número máximo de juicios se llevó a cabo en Maharashtra y el menor número de juicios en el estado nororiental. Entre los estados del noreste, no se llevaron a cabo juicios en Nagaland.7
Figura 2: Distribución estatal de ensayos clínicos en la India (datos de 2007-2015)7
Reactivación del escenario clínico y regulatorio
En 2014, la CDSCO constituyó 12 nuevos comités asesores sobre medicamentos (NDAC) y 25 comités de expertos en la materia (SEC). Estos comités cuentan con varios expertos de eminentes colegios e instituciones gubernamentales para acelerar los plazos de aprobación de un ensayo clínico a 6 o 7 meses.
El proceso de tres niveles consta de:9
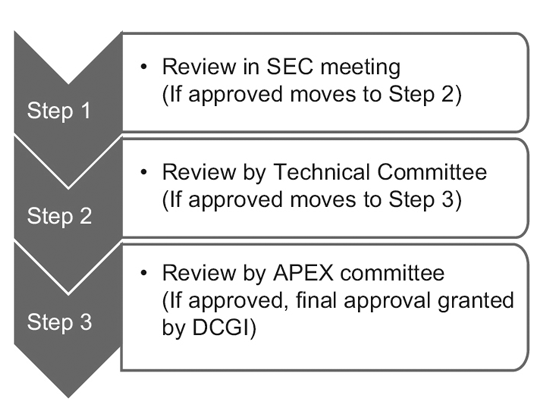
Sin embargo, solo la SEC revisa las solicitudes de ensayos clínicos globales y no se requiere aprobación adicional del comité técnico ni del comité Apex. Las solicitudes de nuevos medicamentos en investigación (IND) también son revisadas de forma independiente por el comité IND y no requieren la aprobación del comité Apex.
Un comité técnico entra en escena sólo si la SEC ha rechazado la solicitud de un patrocinador y el patrocinador se siente agraviado por la decisión. En tal caso, si el comité técnico no está de acuerdo con la decisión de la SEC, tiene el poder de anular la decisión de la SEC.10
En marzo de 2019, el Ministerio de Salud y Bienestar Familiar de la India publicó la Nuevos medicamentos y reglas de ensayos clínicos 2019 con la intención de acelerar la aprobación de ensayos clínicos, nuevos medicamentos, estudios de bioequivalencia (BE) o biodisponibilidad (BA).
Estas normas también han abordado cualquier ambigüedad que existiera respecto a la regulación del Comité de Ética (CE).11
Aspectos destacados de las reglas de ensayos clínicos y nuevos medicamentos, 201911 |
Normas y reglamentos actualizados.11 |
Cronograma de aprobación de ensayos clínicos |
90 días hábiles desde la recepción de una solicitud para medicamentos descubiertos fuera de la India y 30 días hábiles para medicamentos nuevos o IND en la India |
Fabricación de nuevos medicamentos o estudios IND, BE y BA o análisis o exámenes de pruebas. |
Se requiere permiso de la Autoridad Central de Licencias (CLA) |
Renuncia a ensayos clínicos locales |
· Si CLA ha aprobado la comercialización del nuevo medicamento en otros países o ha otorgado permiso para realizar ensayos clínicos globales para el nuevo medicamento en India · No hay evidencia de una diferencia en el metabolismo, la seguridad o la eficacia debido a la diferencia en el perfil genético de la población india. |
Período de validez de un ensayo clínico |
2 años a partir de la fecha de emisión por CLA |
Acceso posterior al ensayo a IND o a un nuevo medicamento |
En circunstancias únicas, el medicamento se distribuirá sin costo a los sujetos del ensayo según las instrucciones de CLA, pero el patrocinador no tiene ninguna responsabilidad por el uso del medicamento después del ensayo. |
Reuniones previas y posteriores a la presentación |
Buscar orientación con respecto a las leyes y procedimientos que rigen el proceso de fabricación y la concesión de licencias o permisos. |
Aprobación de ensayos realizados por la CE y registro de la CE |
· Se debe obtener la aprobación del CE de otro sitio de ensayo si un sitio de ensayo no tiene un CE, y el CE debe estar dentro de los 50 km del sitio de ensayo.· El registro de EC aprobado por CLA sigue siendo válido durante cinco años a partir de la fecha de emisión. |
Condiciones que deben cumplirse para la realización de un ensayo clínico |
· Envío de informe de estado trimestralmente o dependiendo de la duración de la prueba para realizar un seguimiento de la inscripción de los sujetos.· Informes online del estado del ensayo clínico cada seis meses a través del portal SUGHAM para saber si el ensayo está en curso, finalizado o finalizado. |
Tarifa por obtener una licencia, un certificado de registro y un permiso para realizar pruebas |
Diferentes estructuras de tarifas según el propósito del ensayo. Tarifa que oscila entre 50,000 y 5,00,000 INR. |
Superar los desequilibrios
Los desafíos de abordar los ensayos clínicos son multifacéticos e implican el cumplimiento del marco regulatorio de manera responsable y ética por parte de las partes interesadas, el gobierno y el sistema judicial por igual.
Seguridad y protección del paciente Debería ser de suma importancia establecer reglas estrictas para:12
- consentimiento informado mediante grabación audiovisual y en un idioma con el que el paciente se sienta cómodo
- Respeto por los antecedentes culturales, sociales, económicos y educativos del paciente.
- notificación oportuna de EAS
Nuevas reglas básicas que pueden abrir la posibilidad de expandir la investigación médica en la India son:13
- aprobación de las propuestas presentadas a la DCGI dentro de los 30 días siguientes a la solicitud, si no hay comunicación de la DCGI
- seguimiento rápido de aprobaciones nacionales
- Reuniones previas y posteriores a la presentación con el comité de expertos para aportar más transparencia al proceso y establecer un cronograma bien definido para la finalización del ensayo.
- compensación de prueba en caso de que el fármaco en investigación produjera EAG/muerte.
Mano de obra competente e infraestructura de última generación. También juegan un papel importante a la hora de atraer empresas patrocinadoras. Las investigaciones han demostrado que, aunque los ensayos de Fase III se están llevando a cabo a gran escala en la India, los ensayos de Fase I parecen estar limitados al país patrocinador.
Esto podría atribuirse a la aprensión del Patrocinador a la hora de adquirir tecnología y mano de obra calificada. Para permitir que se realicen investigaciones autóctonas en la India, es fundamental brindar una exposición adecuada o educación médica continua al personal y acceso a tecnología actualizada para ser reconocido como un país lo suficientemente competente como para realizar cualquier fase de ensayo.9
Igualmente importante es la necesidad de que haya trabajadores sanitarios cualificados disponibles en todo el país para dar cuenta de la distribución desigual de los ensayos clínicos entre los estados.
Concentrar un ensayo en un estado particular podría llevar a conclusiones sesgadas y simplificar demasiado o exagerar una carga de enfermedad o condición. Al brindar acceso a personas de todos los estados para que se unan a un ensayo clínico, no solo minimizamos el sesgo sino que también incluimos a poblaciones étnicas diversas.9
El futuro de las
Con leyes regulatorias positivas, amigables para los pacientes, rápidas y transparentes, la India seguirá creciendo como un centro internacional para probar y desarrollar medicamentos y dispositivos médicos innovadores.
Fuentes
1. Evangeline L, Mounica NVN, Reddy VS et al. Proceso regulatorio y ética para ensayos clínicos en India (CDSCO). La Revista de Innovación Farmacéutica. 2017;6(4):165-9. http://www.thepharmajournal.com/archives/2017/vol6issue4/PartC/6-4-4-176.pdf
2. Lahiry S, Sinha R, Choudhary S et al. Cambio de paradigma en las regulaciones de ensayos clínicos en la India. Revista india de reumatología. 2018, 13: 51, 5.
3. Gogtay NJ, Ravi R y Thatte UM. Requisitos reglamentarios para ensayos clínicos en la India: lo que los académicos necesitan saber. Indian Journal of Anesthesia. 2017 Mar;61(3):192-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5372399/
4. Ramu B, Kumar MS y Ramakrishna N. Escenario regulatorio actual para la realización de ensayos clínicos en la India. Asuntos Regulatorios Farmacéuticos. Acceso abierto. 2015, 4: 2. https://www.researchgate.net/publication/281765214_Current_Regulatory_Scenario_for_Conducting_Clinical_Trials_in_India
5. Burt T, Sharma P, Dhillon S et al. Entorno de investigación clínica en la India: desafíos y soluciones propuestas. Revista de bioética de investigación clínica. 2014;5:6. DOI: 10.4172/2155-9627.1000201
6. Chaturvedi M, Gogtay Nueva Jersey, Thatte UM. ¿Los ensayos clínicos realizados en la India se ajustan a sus necesidades sanitarias? Una auditoría del Registro de Ensayos Clínicos de la India. Perspectivas en Investigación Clínica. 2017;8(4):172-5.
7. http://ctri.nic.in/Clinicaltrials/news/CTRI_Newsbulletin_July-Dec_2017.pdf Consultado el 23 de abril de 2019.
8. Bhave A y Menon S. Entorno regulatorio para la investigación clínica: pasado reciente y futuro esperado. Perspectivas en Investigación Clínica. 2017, 8: 11.6.
9. Aspectos destacados de las reglas de ensayos clínicos y nuevos medicamentos, 2019. Consultado el 23 de abril de 2019.
10. Dan S, Karmakar S, Ghosh B et al. Digitalización de ensayos clínicos en la India: un nuevo paso de CDSCO para garantizar la credibilidad de los datos y la seguridad del paciente. Asuntos regulatorios farmacéuticos: acceso abierto. 2015;4(3): DOI: 10.4172/2167-7689.1000149.
11. https://www.thehindubusinessline.com/news/new-rules-sweeten-the-deal-for-clinical-trials-by-indian-pharma-cos/article26283499.ece Consultado el 23 de abril de 2019.
Cláusula de exención de responsabilidades:
La información contenida en este artículo tiene como único objetivo brindar orientación general sobre asuntos de interés para el uso personal del lector, quien acepta total responsabilidad por su uso. En consecuencia, la información contenida en este artículo se proporciona en el entendido de que los autores y los editores no se dedican a brindar asesoramiento o servicios profesionales.
Como tal, no debe utilizarse como sustituto de la consulta con un asesor competente. Antes de tomar cualquier decisión o realizar cualquier acción, el lector siempre debe consultar a un asesor profesional en relación con la publicación del artículo correspondiente.
Si bien se ha hecho todo lo posible para garantizar que la información contenida en este artículo se haya obtenido de fuentes confiables, Veeda Clinical Research no es responsable de ningún error u omisión ni de los resultados obtenidos del uso de esta información.
Toda la información contenida en este artículo se proporciona "tal cual", sin garantía de integridad, precisión, puntualidad o de los resultados obtenidos del uso de esta información, y sin garantía de ningún tipo, expresa o implícita, incluida, entre otras. a garantías de desempeño, comerciabilidad e idoneidad para un propósito particular.
Nada de lo contenido en este documento sustituirá, en ninguna medida, las investigaciones independientes y el sólido criterio técnico y comercial del lector. En ningún caso Investigación clínica Veeda, o sus socios, empleados o agentes, serán responsables ante el lector o cualquier otra persona por cualquier decisión o acción tomada basándose en la información de este artículo o por cualquier daño consecuente, especial o similar, incluso si se le advierte de la posibilidad. de tales daños.
Ninguna parte de esta publicación puede reproducirse, almacenarse en un sistema de recuperación o transmitirse de ninguna forma o por ningún medio, mecánico, electrónico, fotocopia, grabación o de otro tipo, sin el permiso previo por escrito del editor.
Para información, contáctenos al:
Veeda Investigación Clínica Privada Limitada
Complejo Vedant, al lado del YMCA Club, SG Highway,
Vejalpur, Ahmedabad – 380 051,
Guyarat India.
Teléfono: +91-79-3001-3000
Fax: +91-79-3001-3010
Email: info@veedacr.com


{kind=link}