
인도는 국내 및 국제 임상시험 플랫폼에 기여할 수 있는 엄청난 잠재력을 가진 국가로 떠오르고 있습니다.
XNUMXD덴탈의 중앙의약품표준관리기구(CDSCO) 는 인도 의약품 통제국(DCGI)이 관리하는 인도의 국가 규제 기관입니다.1,2,3
DCGI는 후원자, 제조 단위 및 트레일 현장의 검사 조정을 담당합니다.3
임상 개발 초기
2000년에 인도의학연구협의회(ICMR)는 인간을 대상으로 한 생물의학 연구 수행에 대한 윤리적 지침을 제정했습니다.4 2005년에는 인도 규제법을 국제적으로 인정되는 정의 및 절차에 맞추기 위해 1945년 의약품 및 화장품법의 Schedule Y가 개정되었습니다.
변경 사항은 다음과 같습니다.
- 정의 1 단계 임상 XNUMX상으로
- 후원자(들)와 조사자(들)의 명확한 책임
- 승인된 연구 프로토콜에 대한 편차 또는 변경 사항을 기록하는 옵션
인도는 또한 인도에서 더 많은 임상시험을 실시할 수 있는 가능성을 열어주기 위해 2005년 무역 관련 지적재산권(TRIPS) 협정을 체결했습니다.5
규제법을 국제 표준에 맞추는 것 외에도 인도는 다음과 같은 이유로 임상 시험에 유리한 목적지가 되었습니다.6
- 영어를 구사하는 의료 전문가
- 전문 기술
- 경제 성장
- 세계적 수준의 기술
- 규모가 크고 다양하며 치료 경험이 없는 인구
임상 시험을 위해 다시 설정
규정 변경에도 불구하고 많은 다국적 제약회사는 임상시험에 대한 지식이 부족하거나 문맹인 대규모 인구를 이용했습니다. 또한, 잘못 정의된 의료 시스템으로 인해 비윤리적인 관행을 모니터링하는 데 어려움이 가중되었습니다.
이로 인해 감독이 거의 없이 임상 시험을 수행하고 서면 또는 시청각 콘텐츠로 환자 사전 동의를 기록하지 않았습니다.
알려진 심각한 부작용을 밝히지 않은 채 환자에게 연구용 약물이나 장치를 투여했으며, 일부는 피험자의 사망을 초래했습니다. 더욱이, 환자의 사망이 임상시험용 제품이나 기기와 관련이 있는지 여부를 확인하기 위한 독립적인 조사위원회가 구성되지 않았습니다.4
2010년부터 2013년까지는 어려운 시기였습니다. 인도 임상시험 비윤리적인 재판으로 인해 누적된 폐해로 인한 시나리오.
그러나 더 나은 규제 프레임워크가 마련되면서 인도의 임상 시험 등록부(CTRI)는 그림 1에서 볼 수 있듯이 수행되는 시험 수가 꾸준히 증가하는 것을 기록했습니다. 또한 대부분의 시험이 XNUMX상인 것으로 관찰되었습니다. 시련.7
그림 1: 수년간의 임상시험 동향
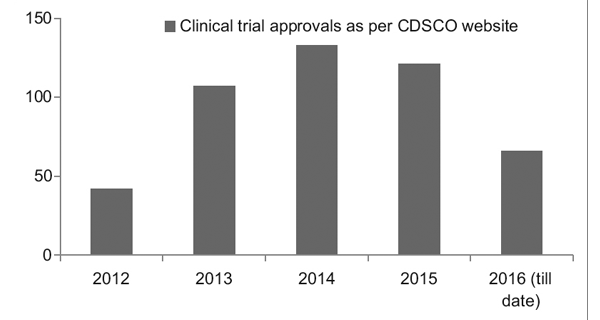
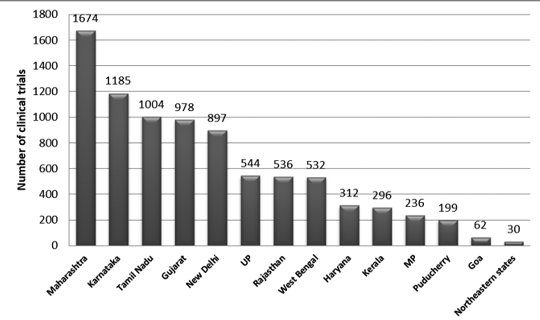
그림 2는 2007년부터 2015년까지 인도의 주별 시험 분포를 보여줍니다. 이 기간 동안 약 3330개의 트레일이 등록되었습니다.
Maharashtra에서 가장 많은 임상시험이 수행되었으며, Northeastern 주에서 가장 적은 임상시험이 수행된 것으로 관찰되었습니다. 북동부 주 중에서 나갈랜드에서는 재판이 실시되지 않았습니다.7
그림 2: 인도 임상시험의 주별 분포(2007~2015년 데이터)7
임상 및 규제 시나리오의 부활
CDSCO는 2014년 12개 신약자문위원회(NDAC)와 25개 주제전문위원회(SEC)를 구성했다. 이 위원회에는 임상 시험 승인 기간을 6~7개월로 단축하기 위해 저명한 정부 대학 및 기관의 전문가들이 다수 포함되어 있습니다.
XNUMX단계 프로세스는 다음으로 구성됩니다.9
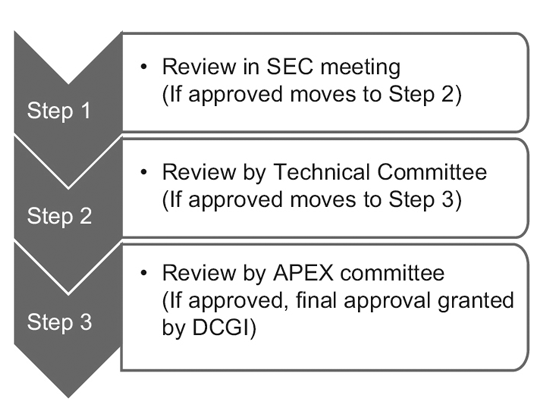
그러나 SEC만이 글로벌 임상시험 신청을 검토하며, 기술위원회나 Apex 위원회의 추가 승인은 필요하지 않습니다. IND(임상시험용 신약) 신청도 IND 위원회에서 독립적으로 검토되며 Apex 위원회의 승인이 필요하지 않습니다.
SEC가 스폰서의 신청을 거부하고 스폰서가 그 결정에 불만을 느끼는 경우에만 기술 위원회가 등장합니다. 이러한 경우 기술위원회가 SEC의 결정에 동의하지 않을 경우 SEC의 결정을 번복할 수 있는 권한을 갖습니다.10
2019년 XNUMX월 인도 보건가족복지부는 신약 및 임상시험 규정 2019년에는 임상 시험, 신약, 생물학적 동등성(BE) 또는 생물학적 이용 가능성(BA) 연구에 대한 승인을 신속하게 처리할 계획입니다.
또한 이러한 규칙은 윤리위원회(EC)의 규정과 관련하여 존재했던 모든 모호성을 해결했습니다.11
2019년 신약 및 임상시험 규정 하이라이트11 |
업데이트된 규칙 및 규정11 |
임상시험 승인 일정 |
인도 외 지역에서 발견된 의약품의 경우 신청서 접수일로부터 영업일 기준 90일, 인도 내 신약 또는 IND의 경우 영업일 기준 30일 |
신약의 제조 또는 IND, BE 및 BA 연구 또는 시험 분석 또는 검사 |
CLA(중앙 라이센스 기관)의 허가가 필요합니다. |
국내 임상시험 포기 |
· CLA가 해당 신약의 타 국가 시판을 승인하거나 인도에서 해당 신약에 대한 글로벌 임상시험을 허가한 경우 · 인도 인구의 유전적 특성의 차이로 인해 신진대사, 안전성 또는 효능에 차이가 있다는 증거는 없습니다. |
임상시험의 유효기간 |
CLA 발행일로부터 2년 |
IND 또는 신약에 대한 시험 후 접근 |
특수한 상황에서 CLA의 지시에 따라 약품은 시험 대상자에게 무료로 배포되어야 하지만, 시험 후 약품 사용에 대한 책임은 의뢰자에게 없습니다. |
제출 전 및 제출 후 회의 |
제조, 라이센스 또는 허가 부여 과정을 규율하는 법률 및 절차에 관한 지침을 구합니다. |
EC가 실시한 임상시험 승인 및 EC 등록 |
· 시험 장소에 EC가 없고 EC가 시험 장소로부터 50km 이내에 있어야 하는 경우 다른 시험 장소의 EC로부터 승인을 받아야 합니다.· CLA 승인 EC 등록은 발행일로부터 XNUMX년간 유효합니다. |
임상시험 수행을 위해 충족해야 할 조건 |
· 분기별 또는 피험자 등록 추적을 위한 임상시험 기간에 따라 상태 보고서 제출· SUGHAM 포털을 통해 XNUMX개월마다 임상시험 상태를 온라인으로 보고하여 임상시험이 진행 중인지, 완료되었는지, 종료되었는지 알 수 있습니다. |
면허증, 등록증, 재판허가 취득 수수료 |
평가판의 목적에 따라 수수료 구조가 다릅니다. 수수료 범위는 INR 50,000 ~ 5,00,000입니다. |
격차를
임상시험을 다루는 과제는 다면적이며 이해관계자, 정부 및 사법 시스템 모두가 책임감 있고 윤리적인 방식으로 규제 체계를 준수하는 것을 포함합니다.
환자 안전 및 보호 다음에 대한 엄격한 규칙을 세우는 것이 가장 중요합니다.12
- 환자가 편안하게 사용할 수 있는 언어로 시청각 녹음을 통한 사전 동의
- 환자의 문화적, 사회적, 경제적, 교육적 배경을 존중합니다.
- 적시에 SAE 보고
새로운 기본 규칙 인도에서 의학 연구를 확장할 가능성을 열어줄 수 있는 것은 다음과 같습니다.13
- DCGI로부터 연락이 없는 경우, 신청 후 30일 이내에 DCGI에 제출된 제안서 승인
- 국내 승인의 빠른 추적
- 전문가 위원회와의 제출 전후 회의를 통해 프로세스의 투명성을 높이고 임상시험 완료를 위한 잘 정의된 일정을 설정합니다.
- 연구용 약물로 인해 SAE/사망이 발생한 경우의 재판 보상.
숙련된 인력과 최첨단 인프라 스폰서 기업을 유치하는 데에도 중요한 역할을 합니다. 연구에 따르면 인도에서는 XNUMX상 임상시험이 대규모로 진행되고 있지만 XNUMX상 임상시험은 후원 국가에만 국한된 것으로 보입니다.
이는 스폰서가 자격을 갖춘 인력과 기술을 확보하는 데 대한 우려 때문일 수 있습니다. 인도에서 토착 연구가 이루어질 수 있도록 하려면 직원에게 적절한 노출이나 지속적인 의학 교육을 제공하고 최신 기술에 대한 접근을 제공하여 모든 단계 시험을 수행할 수 있는 능력이 있는 국가로 인정받는 것이 중요합니다.9
마찬가지로 중요한 점은 주 전역에 걸쳐 임상 시험이 고르지 않게 분포되어 있는 상황을 설명하기 위해 숙련된 의료 종사자를 전국적으로 활용할 수 있어야 한다는 것입니다.
특정 주에 대한 임상시험의 집중은 편향된 결론으로 이어질 수 있으며 질병 부담이나 상태를 지나치게 단순화하거나 과장할 수 있습니다. 모든 주의 사람들이 임상 시험에 참여할 수 있도록 함으로써 편견을 최소화할 뿐만 아니라 다양한 인종 인구도 포함시킵니다.9
미래
긍정적이고 환자 친화적이며 신속하고 투명한 규제법을 통해 인도는 혁신적인 의약품 및 의료 기기를 테스트하고 개발하는 국제 허브로 계속 성장할 것입니다.
지우면 좋을거같음 . SM
1. 에반젤린 L, 모니카 NVN, 레디 VS 외. 인도의 임상시험에 대한 규제 프로세스 및 윤리(CDSCO). 제약 혁신 저널. 2017;6(4):165-9. http://www.thepharmajournal.com/archives/2017/vol6issue4/PartC/6-4-4-176.pdf
2. 라히리 S, 신하 R, 초드하리 S 외. 인도 임상시험 규정의 패러다임 변화. 인도 류마티스 저널. 2018, 13 : 51 - 5.
3. Gogtay NJ, Ravi R, Thatte UM. 인도의 임상 시험에 대한 규제 요구 사항: 학자들이 알아야 할 사항. 인도 마취과 학회지. 2017 Mar;61(3):192-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5372399/
4. Ramu B, Kumar MS 및 Ramakrishna N. 인도에서 임상 시험 수행에 대한 현재 규제 시나리오. 제약 규제 업무. 오픈 액세스. 2015, 4 : 2. https://www.researchgate.net/publication/281765214_Current_Regulatory_Scenario_for_Conducting_Clinical_Trials_in_India
5. 버트 T, 샤르마 P, 딜론 S 외. 인도의 임상 연구 환경: 과제 및 제안된 솔루션. 임상 연구 생명 윤리 저널. 2014;5:6. DOI: 10.4172/2155-9627.1000201
6. Chaturvedi M, Gogtay NJ, Thatte UM. 인도에서 실시된 임상시험이 인도의 의료 요구에 부합합니까? 인도 임상시험 등록소 감사. 임상 연구의 관점. 2017;8(4):172-5.
7. http://ctri.nic.in/Clinicaltrials/news/CTRI_Newsbulletin_July-Dec_2017.pdf 23년 2019월 XNUMX일에 액세스함.
8. Bhave A 및 Menon S. 임상 연구를 위한 규제 환경: 최근 과거와 예상되는 미래. 임상 연구의 관점. 2017, 8 : 11.6.
9. 2019년 신약 및 임상시험 규정의 주요 하이라이트. 23년 2019월 XNUMX일 접속
10. 댄 S, 카르마카르 S, 고쉬 B 외. 인도 임상시험의 디지털화: 데이터 신뢰성과 환자 안전을 보장하기 위한 CDSCO의 새로운 단계. 제약 규제 업무: 오픈 액세스. 2015;4(3): DOI: 10.4172/2167-7689.1000149.
11. https://www.thehindubusinessline.com/news/new-rules-sweeten-the-deal-for-clinical-trials-by-indian-pharma-cos/article26283499.ece 23년 2019월 XNUMX일에 확인함.
부인 성명:
이 기사에 포함된 정보는 독자의 개인적인 사용에 관한 일반적인 지침을 제공하기 위한 것이며, 독자는 해당 정보의 사용에 대한 전적인 책임을 집니다. 따라서 이 기사의 정보는 저자와 출판사가 전문적인 조언이나 서비스를 제공하는 데 관여하지 않는다는 점을 이해하여 제공됩니다.
따라서, 유능한 조언자와의 상담을 대신하여 이용되어서는 안 됩니다. 어떤 결정을 내리거나 어떤 조치를 취하기 전에 독자는 항상 관련 기사 게시와 관련된 전문 조언자와 상담해야 합니다.
이 기사에 포함된 정보가 신뢰할 수 있는 출처에서 얻어졌는지 확인하기 위해 모든 노력을 기울였으나 Veeda Clinical Research는 오류나 누락 또는 이 정보의 사용으로 얻은 결과에 대해 책임을 지지 않습니다.
이 문서의 모든 정보는 완전성, 정확성, 적시성 또는 이 정보의 사용으로 얻은 결과를 보장하지 않고 "있는 그대로" 제공되며, 이를 포함하되 이에 국한되지 않는 모든 종류의 명시적 또는 묵시적 보증 없이 제공됩니다. 성능, 상품성, 특정 목적에의 적합성을 보증합니다.
본 문서의 어떠한 내용도 독립적인 조사와 독자의 건전한 기술적, 사업적 판단을 대체할 수 없습니다. 어떠한 경우에도 그렇지 않습니다. Veeda 임상 연구, 또는 그 파트너, 직원 또는 대리인은 이 문서의 정보에 따라 이루어진 결정이나 조치에 대해 또는 결과적, 특별 또는 유사한 손해에 대해 독자 또는 다른 사람에게 책임이 있습니다. 이는 가능성을 통보받은 경우에도 마찬가지입니다. 그러한 손해의.
본 발행물의 어떠한 부분도 발행자의 사전 서면 승인 없이는 복제되거나 검색 시스템에 저장되거나 기계적, 전자적, 사진 복사, 녹음 등 어떠한 형태나 수단으로도 전송될 수 없습니다.
자세한 내용은 다음 주소로 문의하세요.:
Veeda 임상 연구 개인 제한
Vedant Complex, YMCA 클럽 옆, SG 고속도로,
베잘푸르, 아마다바드 – 380 051,
구자라트 인도.
전화 : + 91-79-3001-3000
팩스 : + 91 - 79 - 3001 - 3010
이메일 : info@veedacr.com


{kind=link}